图的两种表示方法以及与分子文件的关系

图书管理员
图的两种表示方法以及与分子文件的关系
参考:
https://liuyangjun.blog.csdn.net/
https://liuyangjun.blog.csdn.net/article/details/82759650
https://www.cnblogs.com/kaituorensheng/p/5423131.html
通常使用两种不同的方式来表示一张图:
- 邻接矩阵
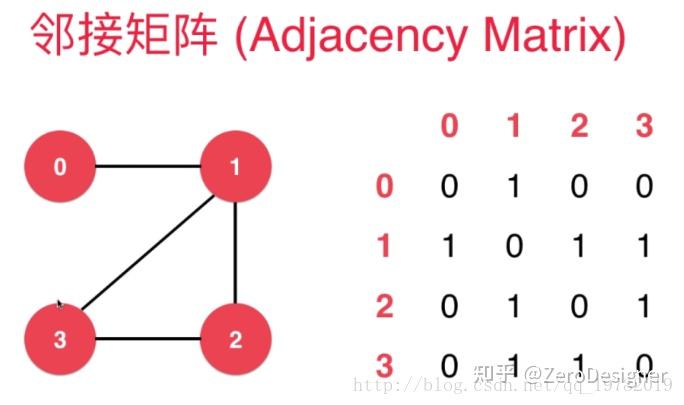
如下图,一张图有4个节点,则对应的邻接表中就有4行4列。
将这个矩阵命名为a,则aij的值代表着i节点与j节点之间是否存在着边,我们用布尔值0和1来表示两种状态,0表示两点之间无连接。1表示两点之间存在着边连接。
因为是无向图,则aij与aji表示的值是一样的.
无向图的邻接矩阵关于斜对角线对称。

- 邻接表
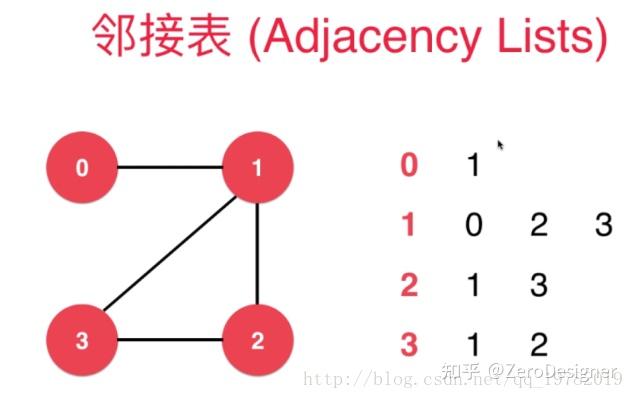
邻接矩阵将所有点与点之间的关系都表示出来,而邻接表则只是把存在关系的点表示了出来。
第0行只有1个1节点,即表示与0节点相连的节点只有1节点,第1行有0,2,3这3个节点,表示着和1节点相连的节点有3个,即0节点,2节点,3节点。第2行后面有1,3节点,表示与2节点相连的节点有两个,分别是1节点和3节点,以此类推......

邻接表表示法也可以用来表示有向图,如下图
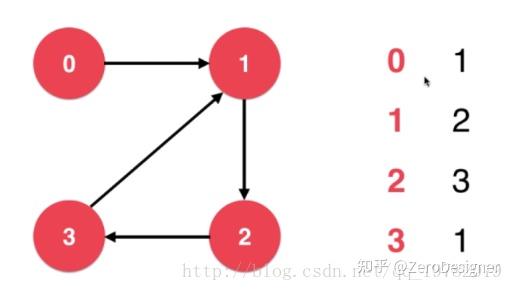

邻接表相比于邻接矩阵来说,所占用的空间更小,这是邻接表的一个优势。但是邻接表如果表示的是一个有很多条边的图,即稠密图的话,则邻接表的优势就不能够完好的体现了。因此,对于一个图来说,我们要根据具体的情况来判断使用哪种方式去表示图,一般邻接表适合表示稀疏图,邻接矩阵适合表示稠密图。
分子格式文件
分子储存文件最主要的是储存其三维坐标以及这些点之间的联系,这里以mol2文件为例
首先看官网介绍:
MOL2格式:http://chemyang.ccnu.edu.cn/ccb/server/AIMMS/mol2.pdf
一个简单的mol2格式文件


我们看@<TRIPOS>BOND这一项,
其格式为:
bond_id origin_atom_id target_atom_id bond_type
- bond_id (integer) =键ID号
- origin_atom_id (integer) = 键初始端原子ID
- target_atom_id (integer) = 键末端原子ID
- bond_type (string) = 键类型
- status_bits (string) = 与键关联的SYBYL状态位。 这些不应该由用户设置。 有效状态值为TYPECOL,GROUP,CAP,BACKBONE,DICT和INTERRES。
Bond Types
- 1 = 单键
- 2 = 双键
- 3 = 三键
- am = 酰胺键
- ar = 芳香键
- du = 虚键(虚设键)
- un = 未知
- nc = 非连接
Example:
5 4 9 am BACKBONE|DICT|INTERRES
5 4 9 am
第一个示例中的键的ID号为5,并连接原子4和9。它是酰胺键。 状态位指示该键是主链的一部分,连接两个残基,并且在创建分子时使用了词典。
第二个示例是相同键的最简表示。
从上述可以看出,mol2中的@<TRIPOS>BOND表示法为邻接表,且为有向图。


用python表示图
要来看一个包,networkx
Web:https://networkx.org/
安装:
pip install networkx
教程:
import networkx as nx
#创建图
G = nx.Graph()
#加一个节点
G.add_node(1)
#从列表中增加节点
G.add_nodes_from([2, 3])
#查看
In [4]: G.nodes
Out[4]: NodeView((1, 2, 3))
#加一条边
G.add_edge(1, 2)
e = (2, 3)
G.add_edge(*e) # unpack edge tuple*,这步不懂,希望有大佬可以解释一下
#从列表中加边
G.add_edges_from([(1, 2), (1, 3)])
#查看
In [6]: G.edges
Out[6]: EdgeView([(1, 2), (2, 3)])
#为点,边,图增加属性
#为图增加属性
G = nx.Graph(day="Friday")
G.graph
{'day': 'Friday'}
#修改属性
G.graph['day'] = "Monday"
G.graph
{'day': 'Monday'}
#为节点增加属性
G.add_node(1, time='5pm')
G.add_nodes_from([3], time='2pm')
G.nodes[1]
#out
{'time': '5pm'}
G.nodes[1]['room'] = 714
G.nodes.data()
#out
NodeDataView({1: {'time': '5pm', 'room': 714}, 3: {'time': '2pm'}})
#为边增加属性
G.add_edge(1, 2, weight=4.7 )
G.add_edges_from([(3, 4), (4, 5)], color='red')
G.add_edges_from([(1, 2, {'color': 'blue'}), (2, 3, {'weight': 8})])
G[1][2]['weight'] = 4.7
G.edges[3, 4]['weight'] = 4.2
#out
In [21]: G.edges.data()
Out[21]: EdgeDataView([(1, 2, {'weight': 4.7, 'color': 'blue'}), (3, 4, {'color': 'red', 'weight': 4.2}), (3, 2, {'weight': 8}), (4, 5, {'color': 'red'})])所以一张分子图可以表示为
import networkx as nx
#创有向图
#苯环:smiles c1ccccc1
#原子序号: 1 2 3 4 5 6
#原子坐标
@<TRIPOS>ATOM
1 C01 1.405 -0.000 0.000 C.2 1 UNK1 0.000
2 C02 0.702 1.216 0.000 C.2 1 UNK1 0.000
3 C03 -0.702 1.216 0.000 C.2 1 UNK1 0.000
4 C04 -1.404 -0.000 0.000 C.2 1 UNK1 0.000
5 C05 -0.702 -1.216 0.000 C.2 1 UNK1 0.000
6 C06 0.702 -1.216 0.000 C.2 1 UNK1 0.000
#邻接表
@<TRIPOS>BOND
1 1 2 2
2 1 6 1
3 2 3 1
4 3 4 2
5 4 5 1
6 5 6 2
#键,[(1, 2), (1, 6),(2,3),(3,4),(4,5),(5,6)]
GM = nx.DiGraph()
#从列表中增加节点
GM.add_edges_from([(1, 2), (1, 6),(2,3),(3,4),(4,5),(5,6)])
#为节点增加属性
GM.nodes[1]['coord'] = [1.405,-0.000,0.000]
GM.nodes[2]['coord'] = [0.702,1.216,0.000]
GM.nodes[3]['coord'] = [-0.702,1.216,0.000]
GM.nodes[4]['coord'] = [-1.404,-0.000,0.000]
GM.nodes[5]['coord'] = [-0.702,-1.216,0.000]
GM.nodes[6]['coord'] = [0.702,-1.216,0.000]
#输出
In [38]: GM.nodes.data()
Out[38]: NodeDataView({1: {'coord': [1.405, -0.0, 0.0]}, 2: {'coord': [0.702, 1.216, 0.0]}, 6: {'coord': [0.702, -1.216, 0.0]}, 3: {'coord': [-0.702, 1.216, 0.0]}, 4: {'coord': [-1.404, -0.0, 0.0]}, 5: {'coord': [-0.702, -1.216, 0.0]}})发布于 2021-02-12 20:31
