给定一串数据序列,如何判断是否存在(模糊的)重复子序列?有什么相应的算法?


最近在给老师写生物信息学教材,我主要写序列比对的部分,所以看了好多的书和视频课,现在把我总结的这部分贴出来和大家共同学习一下。我这部分的内容主要来自北京大学生物信息学公开课的相关内容。
序列比较的基本概念
首先我们来认识一下什么是序列。序列就是由一段字符组成的字符串。组成核酸序列的字符是ATCG(RNA是AUCG)四种碱基,组成蛋白质序列的字符是氨基酸的单个字母缩写。核酸和蛋白质的序列的储存格式通常为FASTA格式。图1就是一个典型的FASTA文件。第一行是大于号加名称或其他的注释,第二行以后是每行60-80个字母(有些也不一定)。


图1.FASTA文件格式,截图自NCBI网站
相似性(similarity) 序列比较的目的是找相似,应用在从数据库做序列相似性搜索。我们是无法使用肉眼在存储量巨大的数据库中找两条相似的序列的。所以我们需要一定的算法或工具尽可能的快速找到相似的序列。相似的序列往往起源于共同的祖先,他们很可能有相似的结构和相似的生物学功能,因此对于一个已知序列但是位置结构和功能的蛋白质,如果与它相似的序列的结构和功能已知,那么就可以推测出未知蛋白质的结构和功能。简言之,相似的序列意味着相似的结构,相似的结构意味着相似的功能。
同源性(homology)在生物学上指两个或多个生物或序列具有多个祖先,在基因序列层面特别是关于系统发育的层次有时还把同源分为直系同源(ortholog)和旁系同源(paralog),直系同源是指在不同物种中的两个序列来自历史上的共同祖先的同一个序列。旁系同源指的是旁系同源的序列因在种内基因复制(gene duplication)而被区分开(separated):若生物体中的某个基因被复制了,功能改变了,那么这些副本序列就是旁系同源的。因此,旁系同源基因存在于同一个物种。如人的α珠蛋白、β珠蛋白、肌红蛋白为旁系同源,相关基因也旁系同源。直系同源的一对序列称为直系同源体(orthologs),旁系同源的一对序列称为旁系同源体(paralogs)。
同源性和相似性有着千丝万缕的联系,两序列有同源性表示两序列来自共同祖先,如果演化时间不长,变化较少,那么两个序列常常会表现出相似性,反之如果演化时间很长,变化会越来越多,两条序列的相似性可能就会越来越低,甚至无法分辨出来,由于同源性常常能带来相似性,而相似性是我们可以通过测量序列来得到的,所以我们常常通过序列的相似性反推或暗示序列的同源性。
序列比较的指标 我们使用序列一致度(identify)和序列相似度(similarity)来描述序列的同源性。序列一致度指的是两个序列长度相同,则他们对应位置上相同的残基的数目占总长度的百分数即为一致度。序列相似度指两个相同长读的序列,它们位置上相似残基相同数或相同残基数目目占总长度的百分数即为相似度,这需要一个替换积分矩阵(subsitution matrix)来统计。替换记分矩阵(Subsitution Matrix)是反映残基之间相互替换率的矩阵,描述了残基两两相似的量化关系。分为DNA替换记分矩阵和蛋白质替换记分矩阵。如果两条序列有不同的长度,那么我们就需要引入空位罚分制度,将两条不同长度的序列通过引入空位的方式对齐成相同长度,然后计算两条序列的相似度和一致度。
2、DNA序列替换记分矩阵
DNA序列替换记分矩阵主要有三种,分为等价矩阵(unitary matrix),转换—颠换矩阵(transition-transversion matrix)和BLAST矩阵。
等价矩阵:该矩阵是最简单的替换记分矩阵,相同碱基之间的匹配得分为1,不同碱基的替换得分为0。但由于该矩阵不含有碱基的理化信息并且不会区别对待不同的替换,因此在实际的序列比较中很少使用。
| A | T | C | G | |
| A | 1 | 0 | 0 | 0 |
| T | 0 | 1 | 0 | 0 |
| C | 0 | 0 | 1 | 0 |
| G | 0 | 0 | 0 | 1 |
表1. DNA等价矩阵
转换颠换矩阵:转换指的是嘌呤和嘌呤之间的替换,或嘧啶和嘧啶之间的替换。颠换指的是嘌呤和嘧啶之间的替换。在进化过程中,转换发生的频率远比颠换高,为了反应这一情况,该矩阵中通常转换的得分为-1,颠换的得分为-5。
| A(嘌呤) | T(嘧啶) | C(嘧啶) | G(嘌呤) | |
| A(嘌呤) | 1 | -5 | -5 | -1 |
| T(嘧啶) | -5 | 1 | -1 | -5 |
| C(嘧啶) | -5 | -1 | 1 | -5 |
| G(嘌呤) | -1 | -5 | -5 | -1 |
表2. DNA转换颠换矩阵
BLAST矩阵:经过大量的实际比对发现,如果另被比对的两个核苷酸相同时得分为+5,不同时得分为-4,则比对效果更好。
| A | T | C | G | |
| A | +5 | -4 | -4 | -4 |
| T | -4 | +5 | -4 | -4 |
| C | -4 | -4 | +5 | -4 |
| G | -4 | -4 | -4 | +5 |
表3. BLAST矩阵
蛋白质替换记分矩阵
蛋白质替换记分矩阵主要有五种,分为等价矩阵(unitary matrix)、PAM矩阵(Dayhoff突变数据矩阵)、BLOSUM矩阵(blocks substitution matrix)、遗传密码矩阵(genetics code matrix, GCM)和疏水矩阵。
等价矩阵(unitary matrix):这是最简单的替换记分矩阵,其中,相同氨基酸之间的匹配得分为1,不同氨基酸间的替换得分为0,由于该方法不考虑氨基酸之间不同的理化性质,也不会区别对待不同的替换,因此在实际的序列比较中较少使用。
PAM矩阵(Dayhoff突变数据矩阵):PAM矩阵基于进化原理。如果两种氨基酸替换频繁,说明自然界易接受这种替换,那么这对氨基酸替换得分就应该高。PAM矩阵是目前蛋白质序列比较中最广泛使用的记分方法之一,基础的PAM-1矩阵反应的是进化产生的每一百个氨基酸平均发生一个突变的量值(由统计方法得到)PAM-1自乘n次,可以得到PAM-n,即发生了更多次突变。
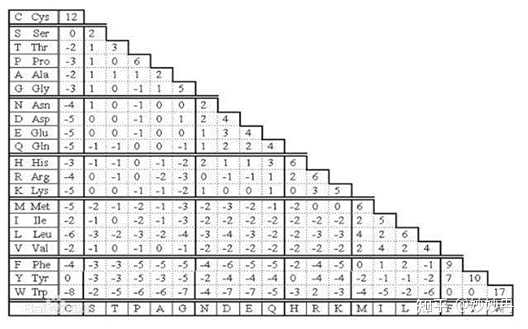

图1.PAM-1矩阵(图片来源百度百科)
BLOSUM矩阵(blocks substitution matrix):BLOSUM矩阵都是通过对大量符合特定要求的序列计算而来的。PAM-1矩阵是基于相似度大于85%的序列计算产生的,那些进化距离较远的矩阵,如PAM-250,是通过PAM-1自乘得到的。可以理解为,BLOSUM矩阵的相似性是根据真实数据产生的,而PAM矩阵是通过矩阵自乘外推而来的。BLOSUM矩阵中的编号,比如BLOSUM-80中的80,代表该矩阵是由一致度≥80%的序列计算而来的,同理,BLOSUM-60是指该矩阵由一致度≥62%的序列计算而来的。BLOSUM-60是最常用的记分矩阵。


图2.BLOSUM-62矩阵(图片来源wiki百科)
遗传密码矩阵(genetics code matrix, GCM):GCM矩阵通过计算一个氨基酸残基转变到另一个氨基酸残基所需的密码子变化数目而得到,矩阵元素的值对应于代价。如果变化一个碱基,就可以使一个氨基酸的密码子改变为另一个氨基酸的密码子,则这两个氨基酸的替换代价为1;如果需要2个碱基的改变,则替换代价为2;以此类推(见表3.4)。注意,Met到Tyr的转变是仅有的密码子三个位置都发生变化的转换。在表3.4中,Glx代表Gly、Gln或Glu,而Asx则代表Asn或Asp,X代表任意氨基酸。GCM常用于进化距离的计算,其优点是计算结果可以直接用于绘制进化树,但是它在蛋白质序列比对尤其是相似程度很低的序列比对中很少被使用。


图3.遗传密码矩阵(图片来源,网络截图)
疏水矩阵:该矩阵是根据氨基酸残基替换前后疏水性的变化而得到得分矩阵。若一次氨基酸替换疏水特性不发生太大的变化,则这种替换得分高,否则替换得分低。

图4.疏水矩阵(图片来源, 网络)
因为DNA序列的变化信息少,在使用简单的DNA替换矩阵比较时,获得的同源性信息远少于蛋白质序列。接下来我们以蛋白质序列比对为例,介绍序列同源性的计算方法。
3、双序列比对网站操作实例:
以Pairwise Sequence Alignment网站为例,我们选择第一个比对工具——Needle,点击进入后,第一步选择要比对的是DNA还是蛋白质,这里我们选择蛋白质。以人血红蛋白的α和β亚基为例,将两条序列输入进去(序列信息可以从NCBI或者Uniprot上获取),点击submit即可开始计算。


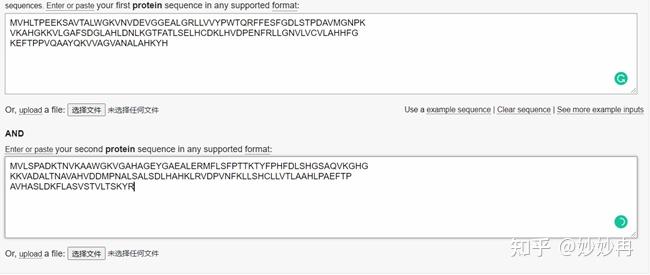

现在来看一下结果。下面一张图展示的是我们刚刚输入进去的两条序列,两条序列比对的中间有三种符合,“|”表示两个氨基酸残基完全一致;’“:”表示性质特别相近的残基,在替换记分矩阵中两两残基替换得分大于1;“.”表示性质微弱相近的残基,在替换记分矩阵中两两残基替换得分小于1。在我们的举例中,这个标记方式是依据BLOSUM-62替换矩阵(默认参数)标记的。通过插入空位(-)将两个序列对齐,空位一般对应的是负分。空位罚分一般由两个部分组成,分为产生一个空位(opening a gap)和空位的延伸(extending a gap),产生一个空位罚分为d分,空位的延伸罚分为e。比如我们图中两条序列比对的第一个空位属于产生一个空位,罚分为d,第二个GA空位属于产生一个空位并延伸一个空位,因此罚分为d+e分,由此可以推导出n个空位的罚分为d+(n-1)*e。将依据替换记分矩阵的打分和空位罚分的得分相加,就得到了最后的两条序列比对得分(如下图所示292.5)。此外,在依据相似性和同源性的计算方法,可以计算出这两条序列的相似度为(60.4)90/146,一致度为43.6%(65/149)。





4、序列比对的算法
前面我们已经介绍了各种替换记分矩阵,利用这些替换记分矩阵,最简单的序列比对方法就是枚举法,这里也叫打分矩阵法(Dot matrix),其次是动态规划模型,最后还有介绍一种我们最常用的Blast算法。
4.1 打分矩阵法
最基本的dot matrix就是把两个序列写在矩阵两边,然后用1标出对应残基相等的位置,如果两条序列完全相等,那么在对角线处可以看到连续的1,如果有局部可以匹配,也会出现对角线连线。Word size是Dot Matrix的唯一参数,如果word size为2表示当对角线出现2个得分时即可算比对成功。
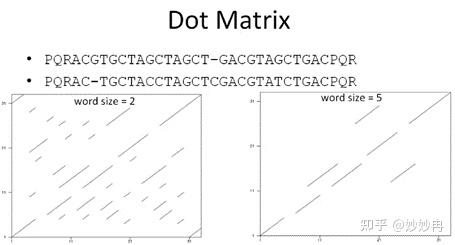

图片来源:视频课截图
4.2 动态规划进行全局比对
利用动态规划模型做序列的全局比对是有上个世纪七十年代芝加哥的两位科学家提出的一种算法,用来寻找全局最优解,该算法也叫做Needleman-Wunsch algorithm。事实上,如果对两条序列做比对,我们又有打分规则,理论上我们可以对两条序列采用枚举法进行比对,最后选择比对分数最大。假设两条长度为n的序列进行比对,那么这两条序列用枚举法需要比对的次数是(2n)!/(n!)2,如果两条序列各长300的话,通过这个公式计算出来的比对次数大约是7*1088,所以枚举法只是在理论上可行,而在实际过程中还需要寻找新的更快的方法。这种方法就是利用动态规划进行全局比对。
从上一节的内容我们已知,一个残基的可能比对方式有两种,一种是对到另外一个残基,另一种是对到一个空位;一对残基的比对有三种可能,一种是两个残基对应,或者是其中一个对应到空位。如下图。此外,我们还知道,最终比对的分数等于各个残基比对的分数之和,所以我们可以得出一个推论是:最好的比对就是之前最好的比对加上当前最好的比对。这就是一个动态规划问题。所谓动态规划就是用来解决具有最优子结构性质的优化问题的计算机算法。而最优子结构性质,就是指局部最优解的组合就是全局最优解。
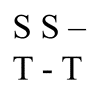
图片自制
动态规划算法的核心思想是把一个复杂的大问题切分为若干个小问题,而后通过逐一找到小问题的最优解,最终解决大问题的最优解。我们用F(i,j)表示x1……i(X序列第1位到第i位)y1……j(Y序列第1位到第j位)。s(A,B)表示残基A和残基B对应的得分,d表示插入空位的得分,则有:
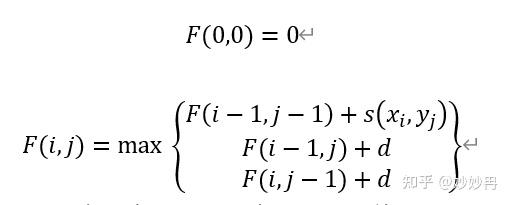

在上面的公式中,我们没有考虑到空位开启和空位延伸的区别。同时,为了便于理解,我们可以把F(i,j)理解成一个矩阵,如下图,那么i,j就可以看做这个矩阵的横纵坐标。这样我们可以更形象的把公式的迭代转换为对矩阵自上而下,自左而右的单元格进行填空。这样的一个过程又可以成为动态规划矩阵。
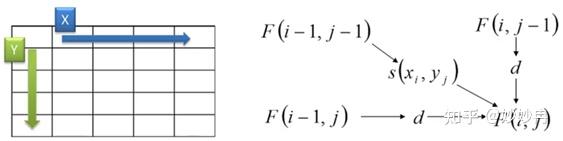

图片来源网络视频截图
4.3算法讲解实例
由于核酸序列由ATCG组成,更简单,所以本实例使用核酸做演示,并使用DNA转换颠换矩阵打分。设d=-7则可以有下图右图填表。
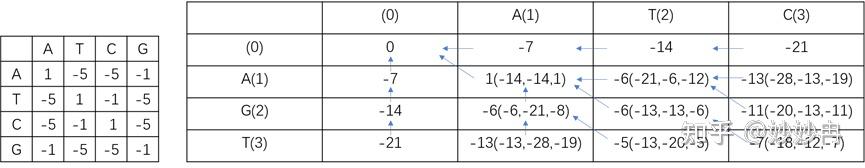

图片自制
除去x1,y1行,剩余的每个空格都有三种得分来源,这个时候我们选择的就是这个位置的最大得分。箭头表示该空格的得分来源。最后我们从F(3,3)点溯源,删除其它信息,那么这一条比对的结果就是全局最优解,因为他们的加和得分也是最大的。比对后序列如图所示。
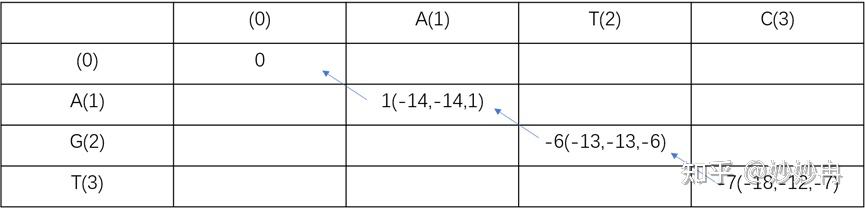

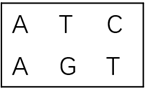
图片自制
4.4局部比对
上一节我们提到的Needleman-Wunsch algorithm算法在早期比对给定的两条序列时受到广泛使用,然而,随着越来越多序列数据的产生,该算法对两条序列所有残基进行比对的算法也遇到了问题。首先,有些功能相近的蛋白,虽然在整体上的序列相似性不高,但是在局部某些功能域序列上会有很高的相似性,这些功能域常常能够独立的发挥作用,但仅靠全局比对是无法发现这些相似的蛋白质,另一方面,在核酸序列比对上,我们需要处理由于内含子导致的大片段的插入缺失造成的比对得分偏差,因而,我们需要新的方法来发现相似的局部序列。
1981年由Smith和Waterman两人提出了一个SmithWaterman算法1解决了这一问题。他们的算法如下:
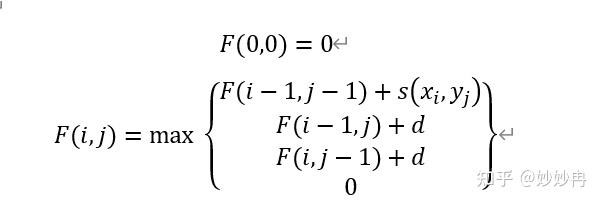

和Needleman-Wunsch的算法相比,他们在F函数中还加入了0。这是在结果中限制了最低分数,也就是0。如果得分为负数,则用0来替换,这样在动态规划矩阵中就不止会出现一条路径,而路径的终点也不一定是右下角,起点不一定是左上角。这样我们可以看到所有的回溯都是局部的,而最终得到的比对也是局部的。也因此,我们除了可以得到最优比对,还可以得到次优比对。
因此局部比对相较于全局比对,通过对比对公式迭代算法的一个简单调整,来引入了一个止损下线,SmithWaterman算法实质上提供了在差异区域扩大之后,重启比对的一个方法,从而可以有效发现局部水平上的相似性。
4.5空位罚分的改进
全局比对和局部比对都没有对空位开始(gap opening)和空位延伸(gap extending)做区分,而是统一使用d来记分,而我们在双序列比对的操作案例中提到,由于空位开始和空位延伸的影响不同,空位罚分的得分往往采取线性组合的模式。如果我们要正确的区分空位开始和空位延伸,就要掌握一个概念——状态。
状态
如前所述,一对残基可能的组合方式有三种,我们称其为三种可能的状态,我们用M,X,Y 表示这三种状态,其中M指两个残基彼此对上,但不一定相等,X表示序列X的残基对到一个空位,或者是在序列X上发生了一次插入,Y表示序列Y的残基对到一个空位,或者在序列Y上发生了一次插入。那么我们就可以用计算机科学中的有限状态机的概念将序列比对描述为在不同状态之间的转移。具体来说,就是从M到X和到Y的转移对应一次gap opening(d),从X或Y到自身的转移对应一次gap extending(e),而从M到自身的转移对应于无空位的情况下比对的延伸s。这样我们实际上就构建了一个马尔可夫链(Markov chain)。马尔可夫链是一个基于概率的随机过程模型,用来刻画一组之间存在关联的随机过程事件,具体来说,马尔可夫链用来描述一组离散状态之间在不同时刻的转移关系。值得注意的是,这里的转移关系不需要是唯一确定的,只需要可以由一个概率分布函数描述即可。
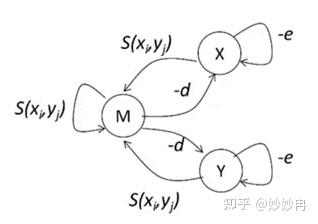
图片来源:网课截图
由此,我们可以写出相应的动态规划改进公式:
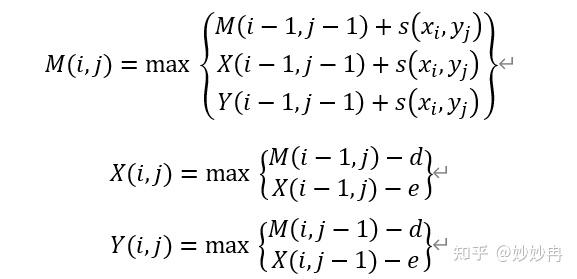

为了更具体的理解这三个公式,我们也可以如动态规划算法一样,将三个公式表示为在三个平面上的动态填充,如下图所示。需要注意的是,对于每一个平面而言,回溯关系既可能来自于本平面,也可能来自于其他平面。
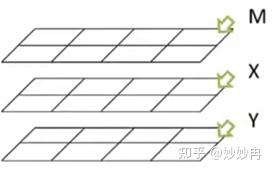
图片来源:视频课截图
4.6 BLAST
BLAST算法自1990年提出2,现今成为序列比对最常用的算法。Blast的核心思想如下:
1、 找到两条序列之间高度相似的小片段,也即种子(seed)
2、 以此为基础,向两端延伸并构建比对。
3、 计算统计显著性,以避免假阳性。
具体来说,blast首先将输入序列切分成若干小段(seed words),而后根据事先建立好的索引表,在数据库中快速定位相关的候选序列,以及在候选序列中的具体位置,通过对所有的seed words进行上述的重复操作,就可以得到查询序列和候选序列之间的heatmap。而最优比对的路径应该平行与主对角线,因此,我们可以进一步去掉那些零散的点,只保留两个或两个以上的片段,将其称为Hit cluster。接下来我们就可以以这些hit clusters为基础,向左右两个方向延伸以扩展,直到总分数的下降超过一个给定的值X之后停止。再扩展后的区域中,我们可以应用动态规划模型算法做比对,从而显著的降低计算量。
为了进一步的提高速度和灵敏度,blast还采取了其它一些技巧。首先blast会屏蔽掉低复杂度的重复性区域,以免产生太多的假阳性hits。低复杂度通常根据序列的信息量(K)来判断,如下公式:
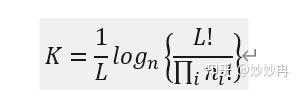
N表示在比对的序列中总共有几种可能出现的字符,核酸为4,蛋白质为20,L表示窗口的长度,n表示各种字符出现的频率。例如,对于一个典型的微卫星序列“CACACACACACACACA”,窗口长度为6,我们可以计算出其K值为0.36,从而屏蔽掉这个序列。
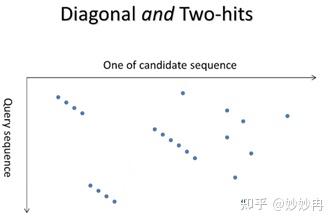
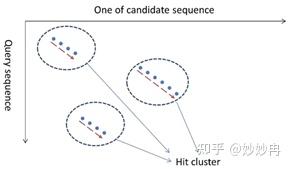
为了提高灵敏度,blast在做第一步seeding的时候除了考虑由查询序列分成的小片段,还会考虑那些与seed words相似的邻居words(neighborhood words),具体来说就是会根据seed words的所有可能变形根据替代矩阵来计算分数。例如以DKT 作为seed word,那么DRT的分数就是13(依据BLOSUM-62),类似的,我们还可以计算出其它neighborhood words的分数,但是为了降低假阳性,我们只选择有高得分的neighborhood words,一般来说要求这个分数要≥11。
BLAST比对质量的评估
在比对之后,还需要评估比对的显著性,以确保这个比对不是由随机因素引起的。在blast中,用E-value对此进行度量。E-value指的是在随机情况下,获得比当前比对分数相等或更高分数的比对条数。例如E-value等于10,则表示会有10个随机匹配获得比当前比对相等或更高的分数。对于E的计算有如下公式:
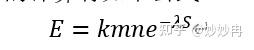
其中m表示要比对的序列长度,n标书数据库的大小,e是自然对数,S是分数,K和λ与打分矩阵相关,用来平衡不同打分矩阵的校正项。由此我们可以得出E值的大小和数据库的大小成正比,也就是说,数据库越大,随机匹配的概率越大。另一方面,E值还有比对的长度成正比,这是因为Blast是局部比对,不需要全长的比对。此外,E值与比对的分数S负相关,也就是说,分数越高,随机匹配的可能性越小。E值在这里表示的是一种期望,并非概率。E值和P值的转换可以用如下公式:
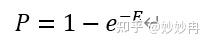
其中e是自然对数。
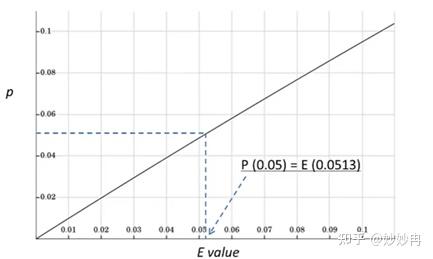

上图是P值和E值的转换关系图,从图中可以看出,当P值等于0.05时,E值也近似的等于0.05。所以有人也将E=0.05作为阈值。
Blast算法区别于动态规划算法的地方是它仅在有限的区域内进行比对,并且并不保证找到全局最优解,它能给出的只是在尽可能快的时间内的最优解。所以Blast的快速运行是以牺牲灵敏度为代价的。
4.7 隐马尔可夫链
在动态规划模型的空位罚分优化中我们借助状态的概念构建了一个马尔可夫链。并简单做了介绍。本节我们将介绍隐马尔可夫链。然而现有的模型只能区分X、M、Y状态,无法区分具体的残基。因此,我们需要进一步引入隐马尔可夫模型,所谓隐马尔可夫模型是指在状态的基础上,增加了符号的概念,每个状态都可以以不一样的概率产生一组观察到的符号,也就是说,除了状态转移概率之外,隐马尔可夫模型进一步引入了生成概率的概念,每个状态都有自己的生成概率分布,可以按照不同的概率产生一组可以被观测到的符号。在隐马尔可夫模型中,状态路径是无法直接看到,这也是“隐”的含义,相反,我们需要根据观测到的符号来推测观测到的状态。