近日,英国女王大学207页博士论文介绍了NLP中的transformer、BERT、预训练模型在蛋白质组学研究中的应用。作者考虑了人工智能和数据驱动分析的情况下,阻碍计算生物学应用中的障碍。在进行这种形式的研究时,有必要考虑所有可能对最终用户有用的应用和实现(如生物标志物研究领域的专家),并要保证所研究的内容必须是新颖的,并与当前的生物学趋势相关,以解决该领域的差距。
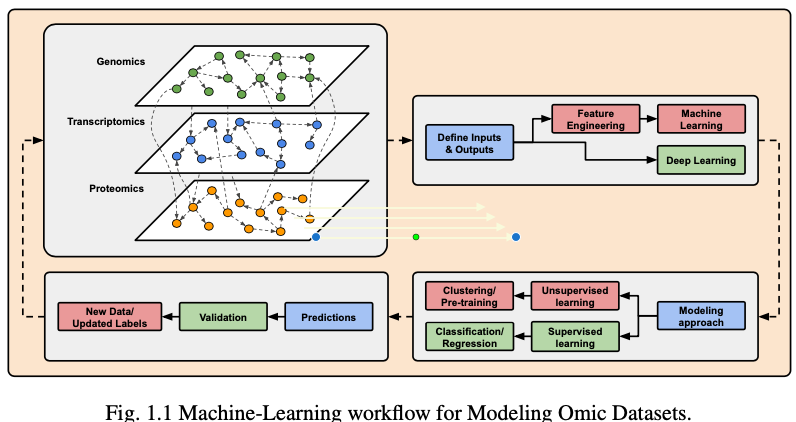
这个工作流介绍了蛋白质组序列分析的领域研究。重点是开发蛋白质序列处理技术,通过深度学习的其他子领域的最先进的方法进行增强建模。
多组学数据分析主要有两个目标
- 了解疾病的发病机制和病因
- 提高我们预测、预防和治疗疾病的能力(即转化医学)。
主要回答如下问题
-
对大量蛋白质组数据的DL模型进行预训练,一旦它被微调到一组下游任务,它的整体性能会提高吗?
-
当只有有限的标记蛋白质组学数据可用时,度量学习能否用于改进微调过程?
-
一旦DL模型被微调到下游任务,它能否用于识别氨基酸序列中与蛋白质功能相关的模式(即基序)?
所有这三个问题在每个章节都会讨论。
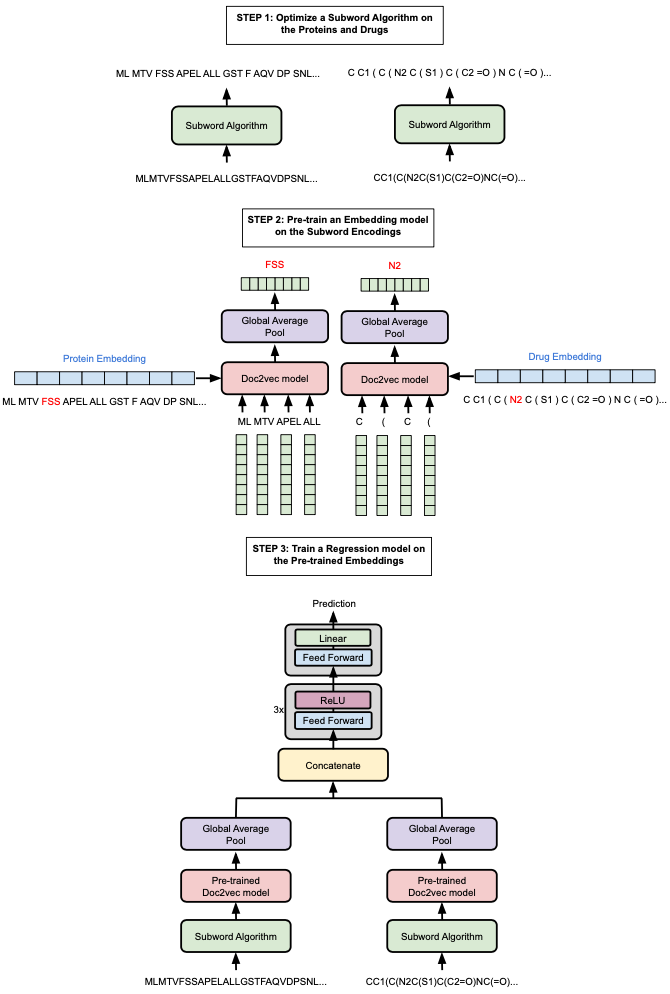
在第三章中,深度学习已被证明是一种建模蛋白质特性的有用工具。然而,考虑到蛋白质长度的多样性,很难有效的总结氨基酸序列。在许多情况下,由于使用固定长度表示法,有关长蛋白质的信息可能会因截断而丢失,或者由于过度填充,模型训练可能会很慢。
作者通过扩展用于表示蛋白质序列的原始词汇表来克服这些问题。具体的,研究了子词算法(Doc2Vec、BPE(Byte-Pair-Encoding))的使用,以生成各种词汇表,并对每种算法产生的预训练编码在多种下游任务上进行了测试:四个蛋白质性质预测任务(质膜定位[plasma membrane localisation]、热稳定性[thermostability]、峰值吸收波长[peak absorption wavelength]、对映体选择性[enantioselectivity])以及在两个数据集上的药物靶点亲和力预测任务。
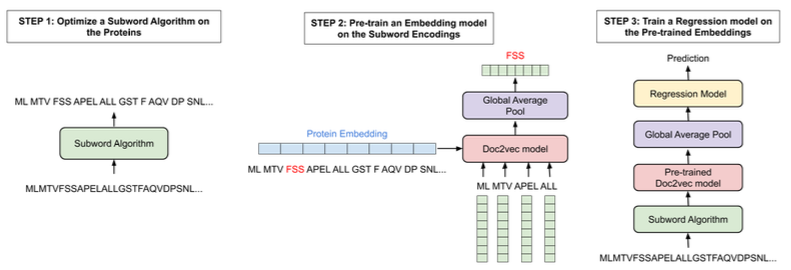
本章中使用的子词算法比之前预训练的Doc2Vec模型提供了更好的整体性能。
在第四章中,作者采用计算机视觉领域的方法,从最少的蛋白质组数据创建一个深度学习模型,将CNN和transformer结合起来,建立了一个最先进的磷酸化位点模型。还利用模型中transformer产生的注意图来识别每个位点中的关键模式。
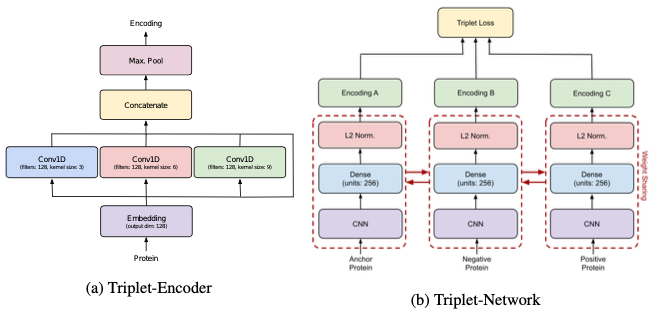
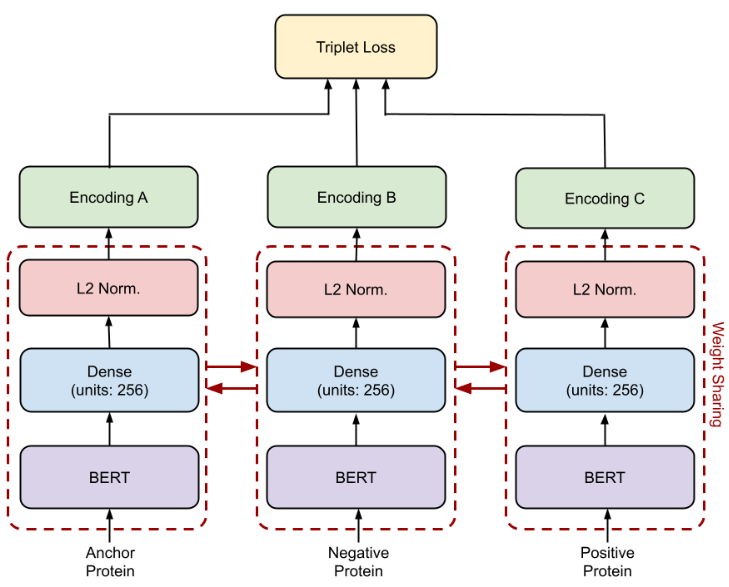
第五章,综合评估了如何应用预训练和度量学习来为一系列下游蛋白质任务开发最先进的结。具体的,作者采用triplet BERT对每个数据集的BERT模型进行微调,并评估其在一组下游任务预测上的性能:质膜定位、热稳定性、峰值吸收波长、对映选择性。
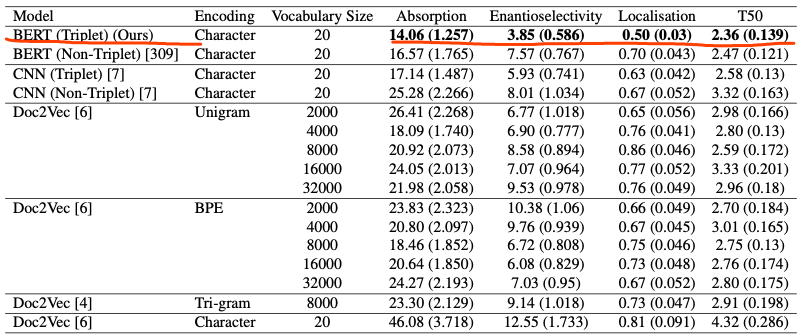
结果显著改善了原始的BERT基线和之前针对每个任务的最先进模型,证明了使用triplet BERT在有限的数据集上优化如此大的预训练模型的好处。
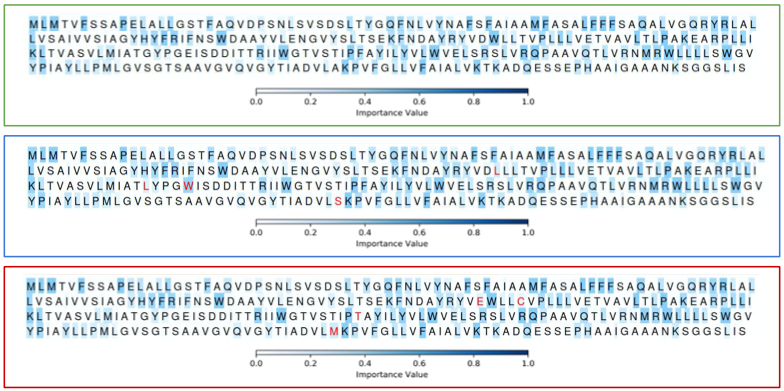
作为一种白盒深度学习,作者还可视化了该模型如何处理蛋白质的特定部分,并检测改变其整体功能的关键修饰。
在最后一章的研究中,作者再次展示了预训练的价值,通过使用两个预训练的BERT模型和一个图卷积网络,为一组药物-靶标相互作用任务生成最先进的结果。
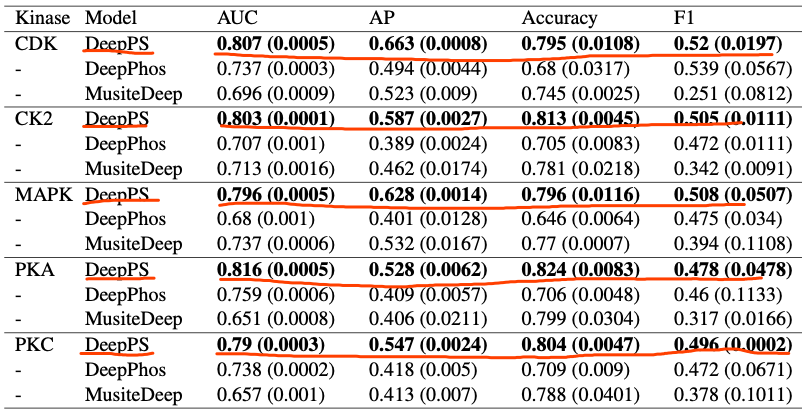
在第六章,深度学习已成为检测蛋白质磷酸化位点的创新工具。然而,负位点和正位点之间的不平衡使得深度学习模型难以准确地对所有位点进行分类。作者通过组合卷积操作和基于transformer的神经网络(下图DeepPS),以形成一个健壮的体系结构,以缓解不平衡位点的影响。
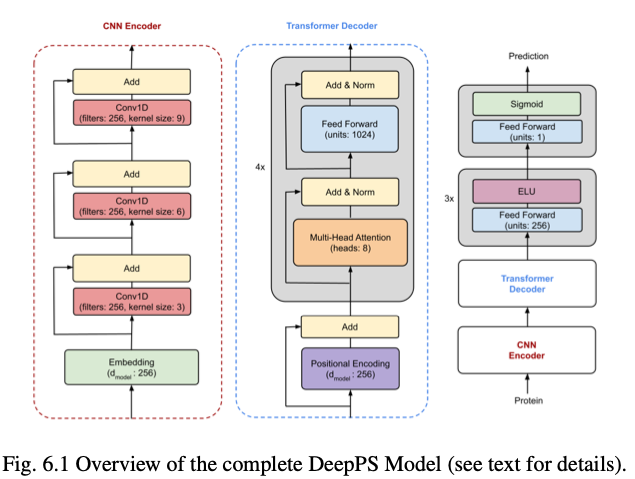
与之前的基线相比,不太可能过度拟合任何一类。
第七章,作者试图利用一组BERT-style的模型,这些模型已经对大量蛋白质和药物数据进行了预训练。
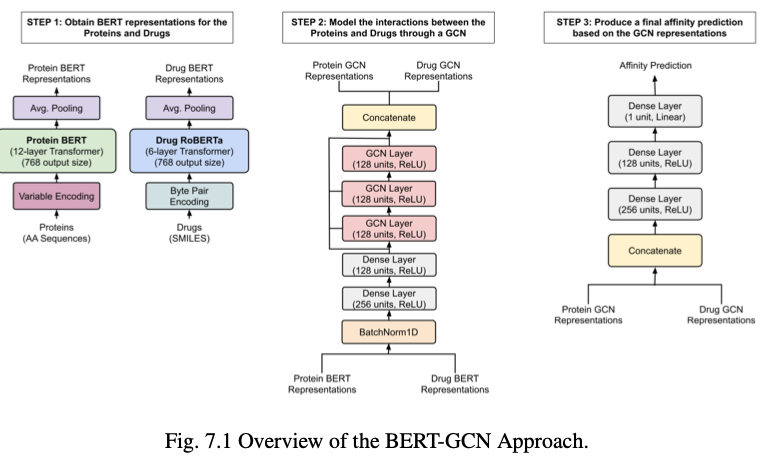
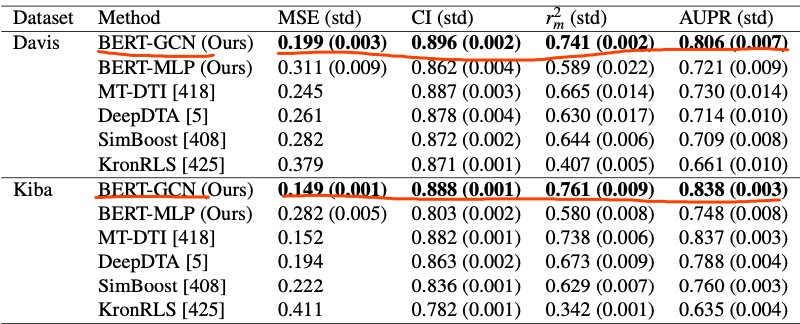
然后,每个模型产生的编码被用作图卷积神经网络的节点表示,而图卷积神经网络又被用于建模相互作用,而无需同时微调蛋白质和药物的BERT模型来完成任务,结果显著改进了基线方法以及每个任务数据集以前的最先进方法。
具体目录如下: