本文介绍一篇来自浙江大学宋明黎教授课题组和侯廷军教授课题组联合发表的一篇文章。该文章提出了一种用于化学反应预测的紧凑的分子字符串表示。该方法基于分子的SMILES字符串表示和Transformer语言翻译模型,通过在预处理阶段对训练集中的输入输出字符串进行对齐操作,来约束输入与输出之间的编辑距离并保证两者的一一对应关系。这使得模型能从学习复杂的SMILES语法中解脱出来,而专注于学习与化学反应相关的化学知识。

1 研究背景 如何高效地设计有效的分子合成路径是药物设计与发现的一个重要研究领域。传统的基于规则的分子合成专家系统往往需要大量的人工编码规则。这种做法不但会受到规则的限制,而且随着反应规则的增加,人工编码规则的成本会越来越高,因此人们开始探索通过计算的方法来预测反应路径。近年来随着人工智能技术的兴起,利用深度学习模型来进行反应预测成为了一种可行的方案。这些反应预测模型大体上可以划分成两类方法:基于选择的以及基于生成的。基于选择的方法将合成预测转化成一个打分或分类问题,其目的是为了尽可能挑出可行的反应模板或目标分子来完成反应预测。这类方法由于在一定程度上引入了编码后的化学知识,更容易完成反应预测,但都无法预测训练集以外的模板或目标分子,这使得其难以泛化到复杂的实际应用中。基于生成的方法则可以缓解这一问题,因此如何提升基于生成的方法的效果成为了一个重要问题。
在当前基于生成的方法中,一种流行的计算模式是先将分子用SMILES字符串表示,再使用Transformer等自然语言翻译模型来将反应预测建模成机器翻译的问题。SMILES(simplified molecular-input line-entry specification)是一种根据分子图的深度优先遍历(depth-first traversal)而生成的分子的字符串表示形式,由于其本身的易读性和易使用性,在反应预测领域得到了广泛应用。由于SMILES是由深度优先遍历而生成的,所以一个分子往往可以通过枚举的方式,来获得多个有效的SMILES表示形式,称之为randomized SMILES。因此,一个化学反应通常也可以用多组不同的输入输出来表示,这作为深度模型的数据增强的一种手段而被许多模型所使用。但因为缺乏输入与输出之间明确的对应关系,这种数据增强实际上隐含了输入和输出之间的一对多关系,这使得计算模型不仅得掌握用于化学反应的相关知识,还得学习复杂的SMILES语法。尽管当前有一些标准化算法(canonicalization algorithm)可以将保证一个分子只能有一个canonical SMILES来表示,但这些标准化算法往往是针对单一分子而设计的,没有考虑化学反应中反应物和生成物的SMILES之间的关系。因此通过这些算法而获得的输入输出SMILES对,尽管保证了输入和输出的一一对应关系,但往往使得输入和输出之间较大的编辑距离(edit distance),最终导致了模型的搜索空间过大;同时这些算法也使得模型无法枚举SMILES来进行数据增强来缓解模型的过拟合问题。
如图1所示,与前人使用的randomized SMILES和 canonical SMILES不同,本研究提出的Root-aligned SMILES(R-SMILES),通过将输入和输出的根原子进行对齐的方式,不仅保证了输入和输出之间的一一对应关系,也大大约束了输入和输出之间的编辑距离,使得两者之间高度相似。这些性质使得模型从学习复杂的SMILES语法中解脱出来,并专注于学习化学反应相关的化学知识。作者将R-SMILES应用到Transformer模型上,在正向反应和逆向反应的多个反应预测任务都进行了实验,都取得了当前最先进的效果。作者还通过可视化Transformer模型中的交叉注意力,来进一步证明模型掌握到了想要的化学反应知识。此外,作者还证明了R-SMILES相比于以往的SMILES表示,在复杂反应(如手性反应)上更加具有优势。最后作者通过让模型预测了多条文献中切实存在的多步反应路径,来阐述在该方法在复杂的现实场景中的应用潜力。
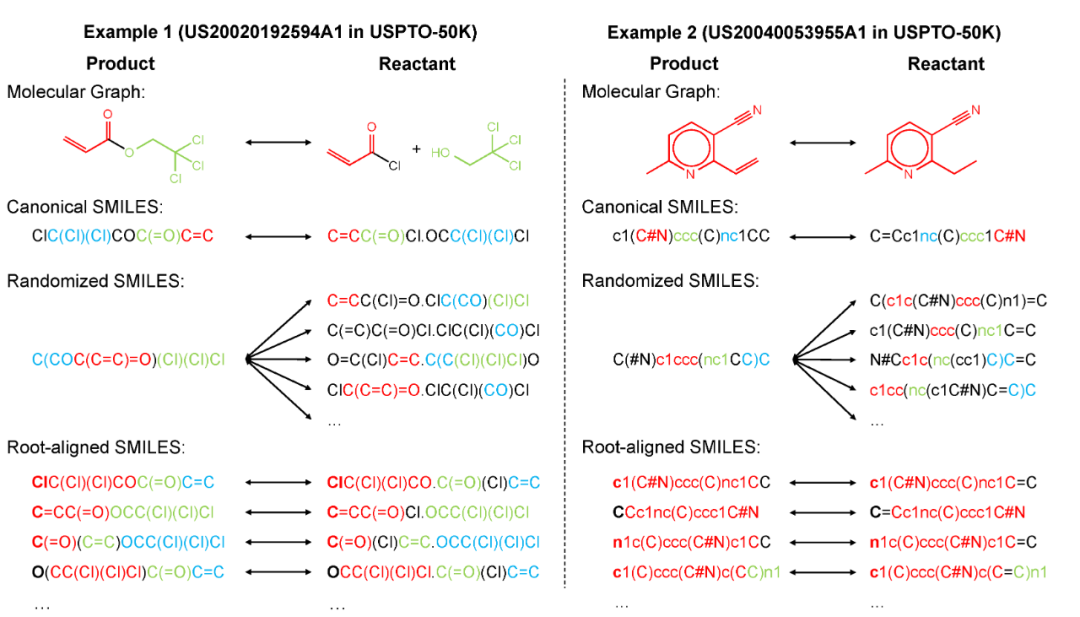
图1. 在逆向反应预测中基于不同的分子表示的输入和输出的对比。
2 实验方法 实验任务 作者在反应物到生成物、生成物到反应物、生成物到合成子、合成子到反应物等多个化学反应预测任务上都进行了实验。为了简化,将反应物(Reactant)简写为R,生成物(Product)简写为P,合成子(Synthon)简写为S,对应的反应预测任务也简写为P2R,R2P,P2S,S2R等。
数据集 本研究中使用了三个公开的分子反应数据集:USPTO-50K,USPTO-MIT与USPTO-FULL,这三个数据集分别包括大约50,000、400,000、1,000,000条反应数据。作者是用了与前人相同的数据划分方式来保证对比的公平性。考虑到现实场景中往往无法获知反应类型,在本研究中进行的所有实验都不包括反应类型的信息。
R-SMILES 在逆向反应的P2R阶段中,获得R-SMILES的流程如表1所示,其中包含以下主要步骤:(1)随机挑选一个带有原子映射的反应SMILES作为原始数据;(2)在生成物SMILES中随机挑选一个原子作为根原子,在表1中挑选了[Cl:8]作为根原子;(3)根据挑选的根原子,获得一个新的生成物SMILES(4)移除生成物SMILES中的原子映射信息。(5)从左往右遍历新的生成物SMILES的原子映射,如果该原子映射在某一个反应物分子的SMILES中出现,那么这个原子映射就作为该反应物SMILES的根原子。在表1中,[C:1]和[Cl:8]被选为两个反应物分子的根原子;(6)根据新的根原子,获得新的反应物SMILES;(7)将(4)和(6)中获得的反应物和生成物的SMILES进行字符划分,获得模型最终的输入和输出。逆向反应的P2S阶段的根对齐操作也与此类似。对于逆向反应的S2R阶段,作者将生成物和合成子拼凑在一起作为模型的输入。为了最小化输入和输出之间的编辑距离,作者将具有一一对应关系的合成子和反应物之间进行根对齐操作,而生成物则是向最大的合成子对齐。在正向反应的R2P阶段,作者将生成物向最大的反应物进行对齐。 表1. 在逆向合成的P2R阶段进行根对齐操作

3 实验结果 使用R-SMILES后的编辑距离 表2. 有无根对齐下的编辑距离比较。Datasetxm::m为数据增强的倍数。Pro.:生成物SMILES。Rea.:反应物SMILES。
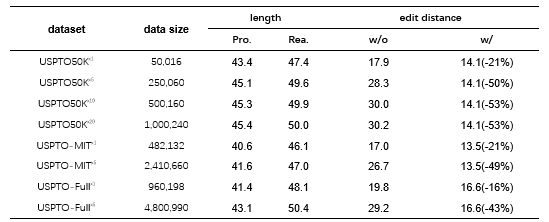
作者首先分析了在有无根对齐操作下,对于逆向预测的P2R阶段的输入输出之间的编辑距离的变化。编辑距离指的是由一个字符串通过增加、修改、删除字符来获得另一个字符串所需要的操作数。可以看到与canonical SMILES对比时(即不进行数据增强的情况),使用R-SMILES后在三个数据集上编辑距离分别下降了21%、21%和16%。而在与randomized SMILES对比时(即进行数据增强的情况),R-SMILES减小编辑距离的效果变得更加显著。在USPTO-50K数据集上进行5倍数据增强时,在使用R-SMILES后编辑距离保持不变,而不使用R-SMILES时编辑距离从17.9增长到了28.3,甚至达到了使用R-SMILES后的编辑距离的两倍以上。
表3. 在USPTO-MIT数据集上R2P阶段的top-K正确率。
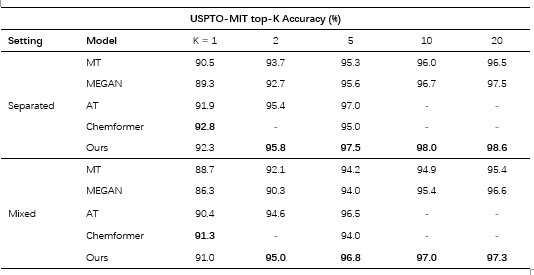
表4. 在USPTO-50K(上)、USPTO-MIT(中)、USPTO-FULL(下)数据集上的P2R阶段的top-K正确率。
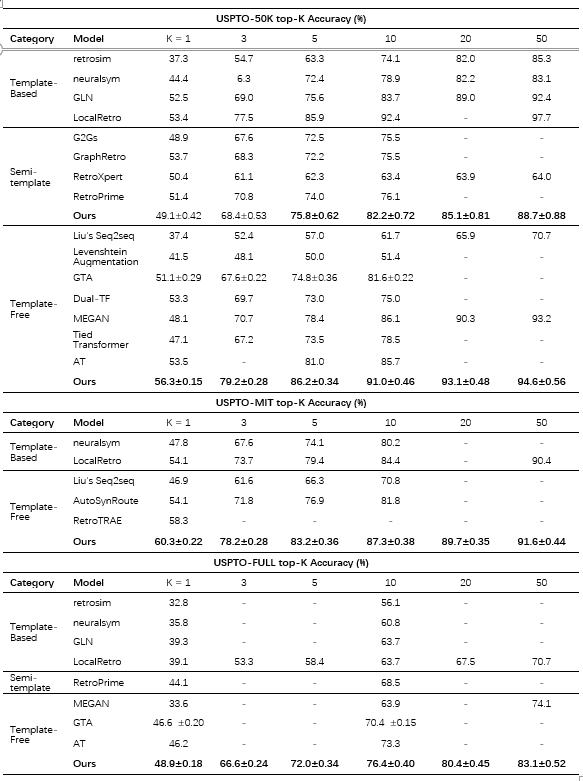
与其他先进方法的比较 作者在这里主要选择了top-K正确率来与其他方法进行比较,top-K正确率代表的是预测的前K个结果中出现正确结果的百分比。
**正向反应预测:**作者在两种不同的实验设置“separated”和“mixed”下进行了实验。这两种实验设置的区别是是否在输入的反应物中区分出试剂。如表3所示,可以看到无论是在哪一种实验设置下,作者所提出的方法在除了top-1之外的所有正确率都取得了最好的效果。
逆向反应预测:作者在三个数据集上都进行了实验。实验证明,在数据量较小的USPTO-50K数据集上,作者提出的R-SMILES的方法在同类别的无模板(template-free)和半模板(semi-template)方法中的多个top-K正确率都达到了最先进的效果,其中无模板的方法甚至取得了与基于模板(template-based)的方法相媲美的效果。在数据量更大的USPTO-MIT和USPTO-FULL数据集上,作者的方法不仅在同类方法中取得了最先进的效果,且也大大优于基于模板的方法。在这里作者指出,在较大的数据集上,基于模板的方法的准确率会随着模板数量增多而下降,同时也无法覆盖测试集上的模板,最终导致了较低的准确率。
注意力可视化 作者进一步展示了在分别使用canonical SMILES和R-SMILES情况下训练出来的模型进行逆合成预测时的注意力可视化的对比。作者随机从测试集中挑选了四个分子的canonical SMILES作为输入,并将Transformer的交叉注意力进行可视化,结果如图2所示。在图2a中,作者指出对于使用canonical SMILES训练的模型,当输入和目标输出是高度相似的情况,模型可能可以捕捉到两者之间的对齐信息并做出正确的预测,但对于每一个输出的字符而言,都需要过分地关注于SMILE语法相关的字符,如‘)’,且这一现象存在于所有基于canonical SMILES所获得的注意力图中。而基于R-SMILES所获得的注意力图则不存在该现象,同时也做出了正确的预测。在图2c中,尽管输入和目标输出是高度相似的,但基于canonical SMILES训练的模型则给出了一张无序的注意力图并做出了错误的预测。作者认为这表示了其捕捉对齐能力的不足。而对于图2e, g中的输入和输出并不相似的反应,基于canonical SMILES训练的模型再次给出了无序的注意力图并且预测失败。而基于R-SMILES的训练的模型,对于这三个反应则成功地给出了有序的注意力图并预测出了想要的R-SMILES。作者认为这些结果都说明了R-SMILES使得模型能够专注于反应的化学知识,最终达到提高模型的预测准确性的目的。
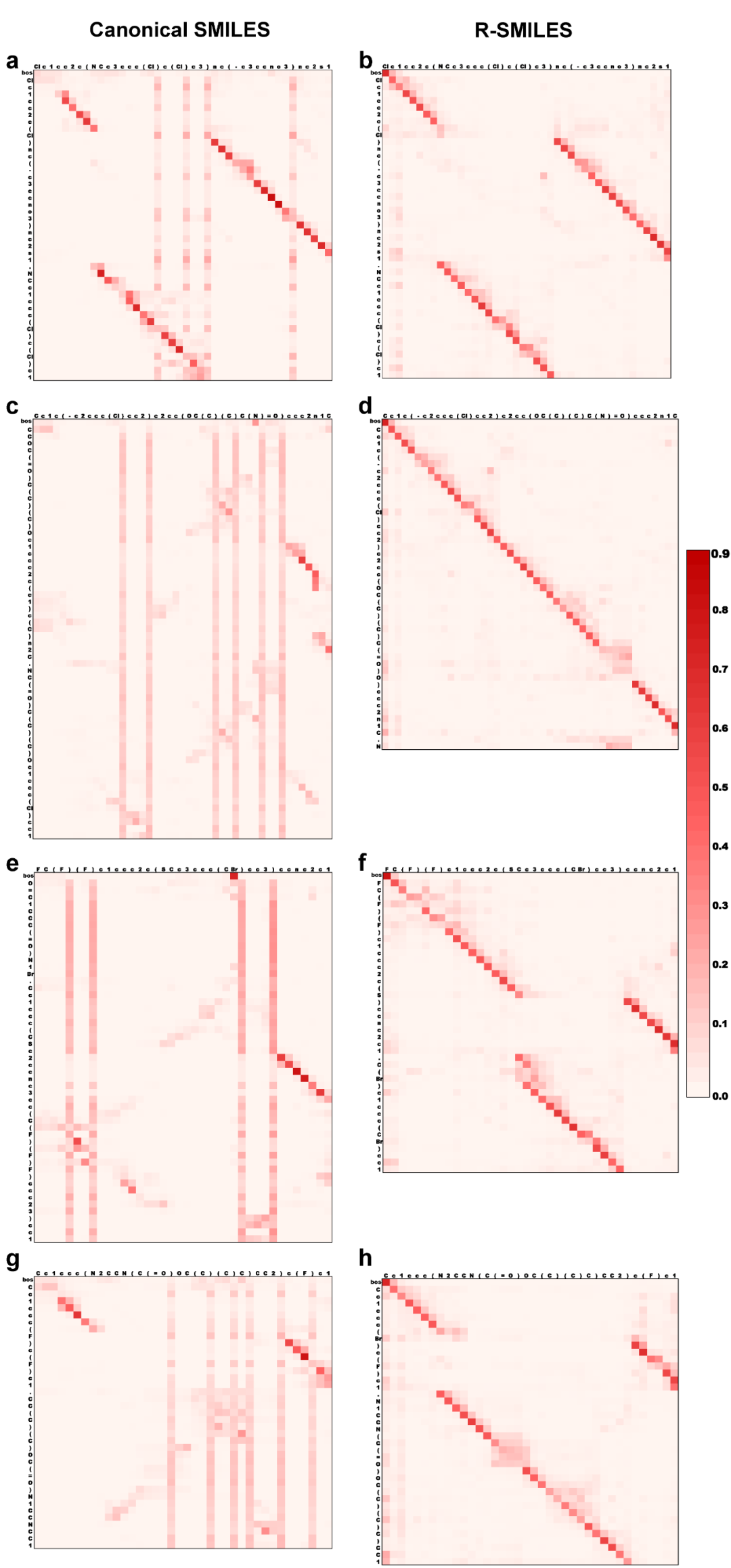
图2. 由canonical SMILES(左)和R-SMILES(右)所获得的注意力可视化。
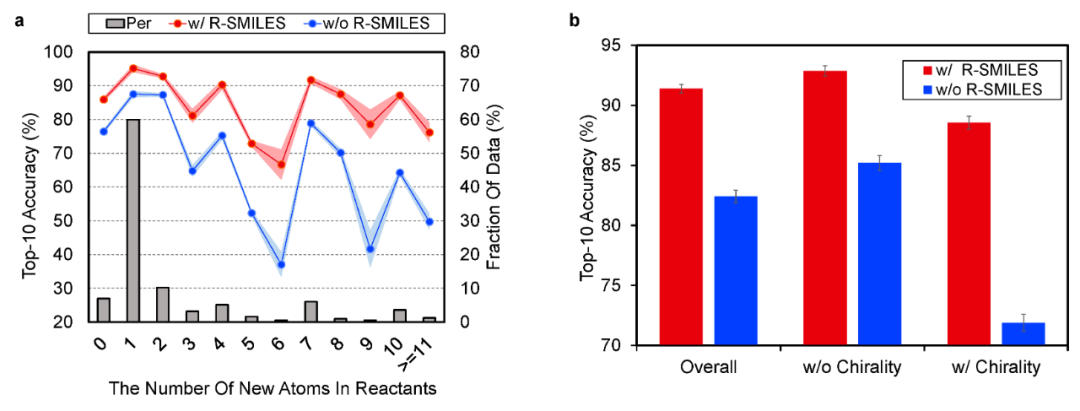
图3. 在复杂反应上的top-10逆向反应预测正确率。(a) 根据新增原子数的正确率;(b)在反应有无手性时的正确率。
对复杂反应的正确率提升 新增原子数量:在图3a中,作者指出,无论新增原子是多少,在使用R-SMILES的情况下总能取得更好的效果,而且这一提升会随着新增原子数量的增多而增大,尤其是对于数据量较少的情况。这是因为R-SMILES减少了输入和输出之间的差异后,模型能够更多地专注于新增的片段。
手性反应:手性是在立体化学中一项重要的分子属性。在图3b中可以看到,在不使用R-SMILES的情况,手性反应的正确率明显低于没有手性的反应的正确率(下降13.3%),而在使用R-SMILES后这一差距大大缩小(下降4.3%)。作者指出R-SMILES在两方面上帮助了手性反应的预测正确率的提升:(1)这是由于手性反应的编辑距离减小的程度更加显著;(2)对于USPTO数据集,进行R-SMILES对齐后的手性标志在反应前后往往是一致的,这大大降低了模型的学习难度。
多步逆合成反应预测 作者通过迭代使用基于R-SMILES训练的逆合成预测模型,成功地复现了多种分子在文献中报告的逆合成路径,进一步验证了方法的有效性。其中对于抗痛风药非布索坦,作者除了复现了文献中报告的逆合成路径之外,还通过模型预测,提出了一条潜在的新的合成路径,如图4a所示。作者分别从产率和价格上分析了这两条路径:(1)作者指出第二条路径中使用的硼酸酯,相比于第一条路径的硼酸有更好的热稳定性,且硼酸频哪醇的引入可以有效地减少副反应的产生,即可以有效地提高产率;(2)在Reaxys数据库中可以发现,第二条路径的原材料相比于第一条路径要便宜得多,因此作者认为他们的方法可能为非布索坦提出了一条更好的合成路径。
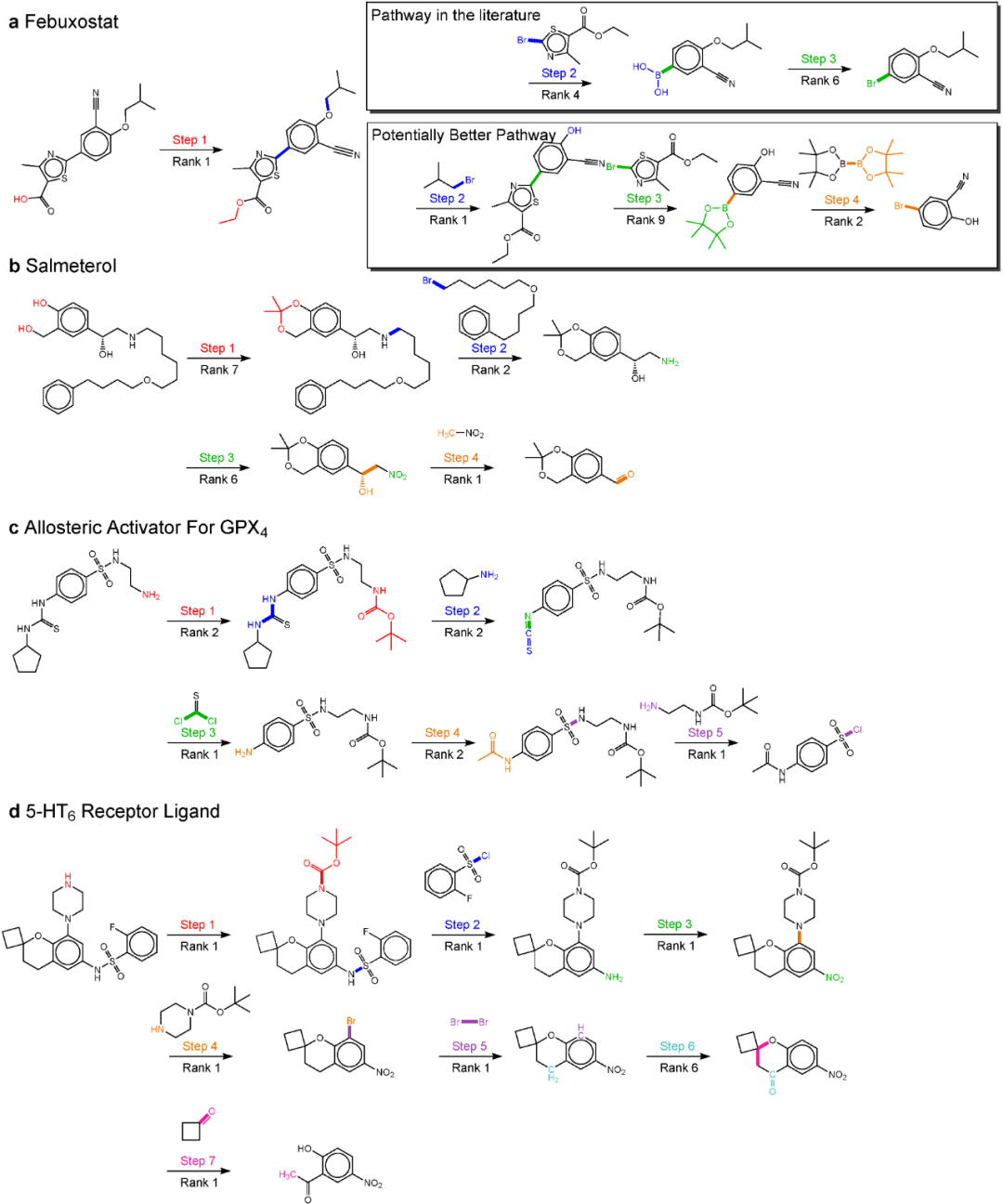
图4. 由R-SMILES训练的模型所提出的多步逆合成预测路径。
4 总结 本文提出了一种用于反应预测的新的分子表示形式R-SMILES,这一方法可以大大缩小输入和输出之间的编辑距离并保证输入和输出之间的一一对应关系。基于这一方法的Transformer模型能在当前主流的公开数据集USPTO上取得最先进的反应预测效果。作者还进一步展示了这一方法在复杂反应、多步逆合成预测上的效果来验证方法的有效性。文章最后作者也指出R-SMILES可以与当前已有的自动化原子映射工具如Indigo、RXNMapper等相结合,应用到没有原子映射的其他数据集上。这些结果表明对于反应预测而言,R-SMILES是一种更为合理的分子字符串表示形式,有望能帮助到相关领域的研究人员。
参考资料 Root-aligned SMILES: A Tight Representation for Chemical Reaction Prediction. Chem. Sci. 2022. DOI: 10.1039/D2SC02763A