2022年6月21日,来自小分子变构药物发现公司HotSpot Therapeutics的Michael Schauperl等人在J Chem Inf Model杂志发表文章,探讨了基于AI的蛋白质结构预测方法对药物发现领域的关键贡献,以及所面临的局限性和挑战。

主要内容整理如下。
摘要 蛋白质是人体的分子机器,其功能失常往往导致疾病。因此,蛋白质是药物发现的关键靶点。蛋白质的三维结构决定了其生物功能,其构象状态决定了底物、辅助因子和蛋白质的结合。合理的药物发现采用工程小分子选择性地与蛋白质相互作用以调节其功能。为了选择性地靶向蛋白质并设计小分子,了解蛋白质的结构及其所有特定的构象至关重要。不幸的是,对于大量与药物发现有关的蛋白质来说,其三维结构还没有通过实验解决。 最近,AlphaFold2,一个基于深度神经网络的机器学习应用,能够以前所未有的准确性预测蛋白质的未知结构。尽管AlphaFold2取得了令人印象深刻的进展,但自然界仍然对结构预测领域提出了挑战。**在本文中,我们探讨了AlphaFold2和相关方法如何帮助提高药物设计效率。**我们强调了先进的机器学习方法在哪些方面需要进一步改进,以便成功地、充分地应用于制药行业。
前言 计算方法和机器学习在药物发现方面有很长的历史。1981年,《财富》杂志宣布了 "下一次工业革命",其中描述了计算机如何帮助药物设计。计算模型几乎存在于药物设计的每个方面。例如,合成可及性和逆合成预测有助于合成规划,大量的序列数据有助于识别新的药物靶点,口袋识别的ML方法是当今最先进的方法,基于ML的毒性预测和PD/PK建模方法也经常被使用。 蛋白质是所有生物系统的分子马达。了解蛋白质及其功能作用对于我们了解生物过程以及药物设计至关重要。 本文探讨了最新一代的机器学习方法如何可能改变蛋白质结构预测的游戏规则,并强调了对药物发现领域的关键贡献。我们讨论了这些方法在复杂的药物设计中所面临的局限性和挑战,并区分了哪些药物设计问题可以用目前的方法 (如RoseTTAFold、RGN2和AF2) 轻松解决,哪些不能。我们为计算生物学家和化学家提供了有效、合理的基于结构的药物设计的要点。我们还展望了在近期和中期内可以对这些算法做出哪些进一步的改进,以利于药物设计。 基于AI的蛋白质结构预测如何帮助基于结构的药物发现 新一代的基于人工智能的结构预测工具已经显示出一些令人印象深刻的成功案例。 CASP访问者之一的Andrei Lupas报告说,来自AF2的结构预测帮助他解决了他的实验室多年来苦苦追寻的一个古细菌跨膜受体的晶体结构。AF2提供了正确的结构模板,可以用来求解实验结构。Baker和同事报道了使用RoseTTAFold解决p101-Gβγ-结合域与PI2Kγ的异源二聚体复合物的冷冻电镜结构。Fowler和Williamson建议使用AF2作为核磁共振结构细化的标准工具。 预测尚未结晶的结构域的蛋白质结构是生物学和药物发现方面的一大飞跃。Pfam是一个广泛使用的基于序列的蛋白质分类资源。到目前为止,四分之三的Pfam结构域家族没有被频繁地结晶 (即每个家族少于10个结构),其中一半以上没有在任何实验结构中发现。正确预测这些结构域使我们能够从结构的角度审视大量的新靶点。可以这么说,如果关于这些蛋白质的唯一知识是其氨基酸序列,那么AF2结构是目前最好的结构。 AF2预测的结构为基于结构的药物设计提供了许多新型蛋白质靶点的入口。三维结构使计算化学家能够在蛋白质表面搜索口袋和功能相关区域。检测一个合适的口袋是药物开发的一个重要步骤。当蛋白质结构已知时,可采用对接、虚拟筛选、自由能计算和其他基于结构的计算方法。 尽管,AF2在所有蛋白质上的表现都不尽相同,但该算法通过提供一种称为预测lDDT得分 (plDDT) 的准确性估计,帮助科学家了解其结构预测的好坏。plDDT得分有助于了解蛋白质的哪些区域被建模为高置信度,因此可以在药物设计过程中以类似于实验结构的方式使用。 **置信度较高的人类蛋白质的预测,对其在药物设计项目中的应用很有意义。**例如,对于F-box LRR重复蛋白-8 (FBXL8_HUMAN),蛋白质数据库 (AF-Q96CD0-F1-model_v1) 中没有具有高序列一致性的模板。然而,AF2可以预测一个具有高置信度的结构 (plDDT=92)。 图2A显示了对人类受体酪氨酸蛋白激酶ERBB-2 (ERBB2) 的预测,并根据其plDDT得分对氨基酸进行着色。ERBB2是曲妥珠单抗的靶点,曲妥珠单抗是一种单克隆抗体,1998年被批准用于治疗HER2+乳腺癌和胃癌。
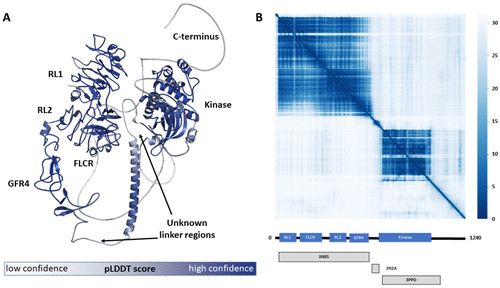
图2. (A) 人类受体酪氨酸蛋白激酶erbB-2蛋白的结构,按预测的lDDT分数着色。(B) ERBB2蛋白的预测对准误差。 不过,激酶结构域相对于其他结构域的方向有一个很大的预测对准误差,因为这个结构域没有像其他结构域那样在同一个晶体中得到解决。如图2A所示,一个长环连接激酶和N端结构域。环路的预测不太可靠,使得方向性预测也不太可靠。这突出表明,准确预测域与域之间的相互作用比预测单域结构更具挑战性。这对药物设计的影响将在下一节进一步讨论。
基于AI的结构化方法的进一步挑战 蛋白质存在多种构象,这些构象都与它的功能作用和生物学影响有关。此外,蛋白质不是仅仅通过其序列就可以完全描述的。体内的蛋白质通常会受到多种翻译后修饰 (PTMs) 的影响,这些修饰会以一种激烈的方式改变蛋白质结构。它们可以与其他蛋白质、辅助因子、DNA和RNA形成复杂的结构,这也可以诱发结构变化。此外,小分子的结合也可以改变蛋白质的结构,例如变构位点。 接下来,我们将讨论蛋白质折叠是如何应用于目前的药物发现方法中的,以及它们的局限性在哪里。
预测所有相关构象状态
解决蛋白质的结构问题通常使药物设计项目向前迈进一大步。然而,蛋白质的实验结构特定于所采用的构建体和蛋白质的PTM状态。此外,一个蛋白质的构象状态是一个集合,单一的代表性结构可能不足以完全描述一个蛋白质及其生物学功能。 AF2由五个模型组成,每个模型都做出了自己的预测。然而,AF2预测的五个不同结构通常非常接近,见图3 (红色结构)。这些相似的预测结构可能无法涵盖一个蛋白质的生物功能的所有方面。
蛋白质确实以许多不同的构象出现,然而药物可能只与蛋白质的某种状态结合。 一个突出的例子是人类钾离子电压门控通道H亚家族成员2 (hERG) 蛋白。 hERG的突变体和小分子与hERG的相互作用可导致先天性长QT综合征。与结构变化相关的动力学非常独特,使其成为重要的蛋白质靶点。hERG K+通道的三个主要构象是关闭、开放和不活跃。AF2对hERG通道的结构预测仅与Cryo-EM预测的结构相似 (见图3A)。除了训练期间提供的构象,AF2不能预测其他相关的构象。
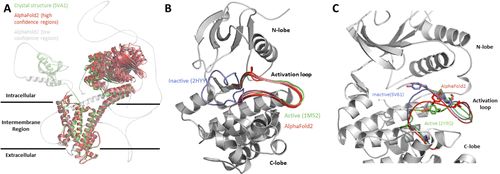
图3:(A) 人类钾离子电压门通道H亚家族成员2的冷冻电镜结构 (5VA1,绿色) 和AlphaFold2预测 (高置信区,红色;低置信区,灰色)。所有的AF2预测都与报道的冷冻电镜结构相似。(B) 人类酪氨酸蛋白激酶ABL1的活性 (1M52,绿色) 和非活性形式 (2HYY,蓝色) 的激活环构象。AF2的预测 (红色) 仅与激活环的活性形式相似。(C) 磷酸化 (活性,绿色,5V61) 、非磷酸化 (非活性,蓝色,2Y9Q) 和AlphaFold2 (红色) 的有丝分裂原激活蛋白激酶-1 (MAPK1) 的激活环构象。AlphaFold2模型预测的是活性和非活性形式的混合。 对于hERG,许多突变和天然变体,以及结合和阻断该通道的化合物都是已知的。关于突变和结构变异的信息直到现在还没有被AF2使用,但经常被制药业的生物学家和化学家用来理解一个蛋白质及其功能。纳入这些突变并理解其结构含义将是未来ML方法的一个良好方向。
另一个例子是人类酪氨酸-蛋白激酶ABL1。 激酶参与多种途径的调节,其激活必须受到严格控制。因此,一个激酶的活性和非活性状态的平衡必须在细胞中得到精确的调节。在一个过于简化的表示中,一个激酶只存在这两种非常不同的构象。虽然绝大多数ATP竞争性抑制剂都与激酶的活性构象结合,但少数小分子,例如抗癌药物伊马替尼,却选择性地与ABL1的非活性形式结合。与特定构象的结合是引入激酶抑制剂的选择性的一种方式。 图3B显示了人类酪氨酸-蛋白激酶ABL1的活性 (开放) 和非活性 (封闭) 构象。然而,**即使激酶的多种构象已被存入蛋白质数据库,AF2也不能预测ABL1的非活性状态。**所有五个AF2模型都预测了激活环的活性构象。从ML的角度来看,这可能是一个理想的结果,因为模型之间的一致性很高,但对于药物设计过程来说是不利的,因为所有的状态都参与了对靶点的调节。 此外,**AF2还不能区分一个预测是对应于一个特定蛋白质的活性或非活性状态。AF2对结构的基本功能没有任何了解,它预测的是它认为最有可能出现在PDB中的状态。**如果对于一个药物设计问题,需要某种状态的结构,那么基于所需构象的相关蛋白质结构建立一个同源模型可能是有利的。 与目前用于AF2的五种模型相比,提出能够预测更多不同构象的模型来覆盖构象差异将是有益的。此外,整合关于不同区域的动力学估计也有助于揭示蛋白质药物相互作用的某些方面。我们想强调的是,研究界已经开始解决这个问题并修改了AlphaFold2。通过引入突变和降低MSA的序列深度,产生了一套更多样化的结构。
预测蛋白质翻译后修饰
结构变化和从活性状态到非活性状态的转变往往与PTMs有关,例如泛素化、磷酸化、乙酰化和甲基化。激酶和磷酸酶是细胞中的核心开关。它们通过对关键残基的磷酸化或去磷酸化来激活或停用某些途径。很多时候,这种PTMs与蛋白质结构的变化相伴而生,如结构域的移动、环路构象的变化、蛋白质的二聚化/聚合。 残基修饰可以引发根本性的结构变化,这在AF2中还没有得到考虑。
在考虑MAPK1时,这一挑战变得很明显。人类MAPK1有活性和非活性构象,这两种构象在PDB中经常被报道 (总共有113个PDB结构)。激活环在残基Thr-185和Tyr-187被磷酸化后改变其构象。有趣的是,AF2预测了两种状态之间的中间构象 (见图3C,红色),因为相应的模型是在所有113个结构上训练的。它不能区分磷酸化、活性 (图3C,绿色) 和非磷酸化、非活性 (图3C,蓝色) 的蛋白质形式。 这种行为的另一个例子是干扰素调控因子 (IRF) 家族。IRFs是特别有趣的药物开发靶点,因为它们参与了肿瘤的抑制和对病原体的免疫反应。IRF蛋白拥有两个不同的结构域:一个N端DNA结合结构域和一个C端调节结构域。C端调节结构域包含一个IRF关联结构域 (IAD),它对二聚体的形成至关重要。IRF通过阻断IAD结构域而具有自抑制功能。磷酸化后,自抑制 (单体) 形式不稳定并形成二聚体,已观察到异二聚体和同二聚体。 出于药物发现的目的,全面了解参与IRFs激活的结构机制是至关重要的。然而,像磷酸化这样的PTM没有包含在目前基于人工智能的结构预测工具的输入序列中,这使得我们无法预测它们因PTM而引起的结构变化。
图4A显示了IRF3的野生型和拟磷突变体的晶体结构和预测的AF2结构。AF2结构总是类似于单体 (无活性、未磷酸化的结构)。尽管PDB中报道了IRF3的一个二聚体结构,但单体结构的发现更为频繁。对于IRF5,ML预测总是产生IAD结构域的非抑制性构象,见图4B。PDB中报告的唯一IRF5结构是二聚体形式。 这个例子强调了AF2预测的结构最有可能在PDB中找到。AF2只能预测PDB中报道的IRF蛋白构象之一。然而,令人惊讶的是,鉴于IRF3和IRF5的折叠结构非常相似,不同的构象被预测出来,尤其是AF2对新的折叠结构有很好的概括性。
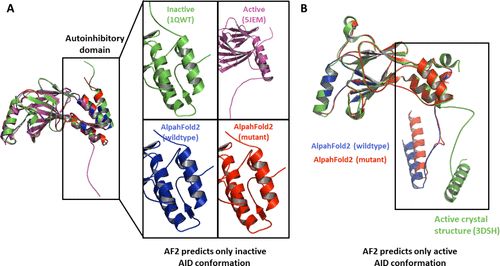
图4:(A) IRF3的单体 (绿色,1QWT) 和二聚体 (粉色,5JEM) 的晶体结构以及AF2对野生型序列 (蓝色) 和IRF3的拟磷突变体S386/396E (红色) 的预测。(B) 二聚体的晶体结构 (绿色,3DSH) 和IRF5的野生型 (蓝色) 拟磷突变体S435/446E (红色) 的AF2预测。预测类似于在 PDB (训练数据) 中发现的单个蛋白质的大多数结构,而不是蛋白质的磷酸化状态。 包括有关PTM及其对结构的影响的信息是一个更难实现的目标。然而,由于PTM引起的结构变化是一个重要因素,基于这种因素修改结构预测方法将显著推进药物设计过程。
预测多域结构
PDB结构并不能完全代表人类的蛋白质组。如图5A所示,某些蛋白质家族和超家族的代表性过高,而其他家族的代表性不足。这意味着,**AF2反映了一个固有的数据偏差,因为它是在PDB上训练的。**众所周知,PDB偏向于容易结晶的蛋白质和单域结构。在所有UniProt家族中,超过40%的家族没有一个晶体结构被报道。 PDB数据库本身和AF2使用的数据扩充都是偏向于单域预测的。这使得AF2在结构域的预测上很准确。然而,大多数人类蛋白质由一个以上的结构域组成。为了理解一个蛋白质的功能,需要准确地捕捉结构域-结构域方向性信息。域间建模是一项更难的任务,因为在PDB中可以学习的合适的例子比在域的层面上要少得多。尽管AF2对多域蛋白质的结构预测不如在域层面上准确,但AF2仍然在CASP14中赢得了这场比赛。 值得注意的是,具有最高置信度的AF2模型并不总是与实验结构具有最佳一致性的模型。例如,在CASP14的T1024挑战中,DeepMind团队提交的第3个模型是正确得到域-域方向的模型 (RMSD Cα = 2.1),模型1的RMSD Cα为5.6,模型2的RMSD Cα为5.7。 对于基于结构的药物设计来说,这些类型的不规则性可能是一个重要的问题,因为它表明所有模型都必须被处理和考虑,这是不实用且昂贵的。
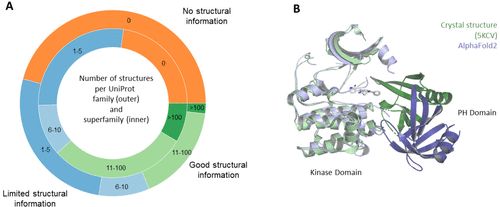
图5:(A) 在PDB中发现的每个UniProt家族 (n = 3892) 和UniProt超家族 (n = 331) 的蛋白质结构数量。70%以上的UniProt家族和50%以上的超家族的实验结构都少于5个。(B) Miransertib (灰色) 与AKT1 (绿色) 的PH和激酶域之间的口袋结合的晶体结构。与晶体结构 (深绿色) 相比,AF2 (蓝色) 预测PH域 (深蓝色) 的方向不同。 针对结构域-结构域相互作用区域的小分子很重要,使制药行业能够追求新的靶点。
Miransertib是一个特别有趣的例子,它是一种与两个结构域形成的口袋结合的化合物。它是一种AKT1的小分子抑制剂。它与激酶和PH结构域之间形成的一个口袋共价结合。 尽管文献中已经报道了结构域-结构域的相互作用,但AF2预测PH结构域的方向与激酶结构域不同 (图5B)。对于新的蛋白质,不能指望域-域的方向平均比已知结构的方向好。为了开发像miransertib这样的药物,对多域结构进行有把握的预测是至关重要的,特别是对域之间形成的口袋。 **提高域与域之间相互作用的准确性是基于人工智能的结构预测工具的未来挑战之一,这可以帮助药物发现。**多个研究小组已经试图通过将AlphaFold2与其他工具相结合或试图改进AF2算法本身来应对这一挑战。然而,在多域预测的准确性与单体预测相当之前,必须进一步改进应用,以可靠地预测多域蛋白质。
预测蛋白质-蛋白质/RNA/DNA复合物
在IRFs中,结构的变化使异质或同质复合物的形成成为可能。复合物的形成是自然界中广泛存在的现象,针对复合物的药物也相当普遍。蛋白质-蛋白质相互作用 (PPI) 是当今化学生物学和药物发现的主要挑战之一。 然而,有几种与蛋白质-蛋白质界面结合的药物已经上市了。Tafamadis针对的是转甲状腺素四聚体形式的两个甲状腺素结合点中的一个。该小分子稳定了与家族性淀粉样多发性神经病 (FAP) 相关的四聚体形式的蛋白质,如图6A所示。在FAP中,转甲状腺素单体没有紧密结合,可以从四聚体上脱落,错误折叠,然后聚集。这些聚集物对神经系统造成伤害。预见这种多域的相互作用将有助于合理的药物设计,以便在未来开发出像Tafamadis的小分子抑制剂。
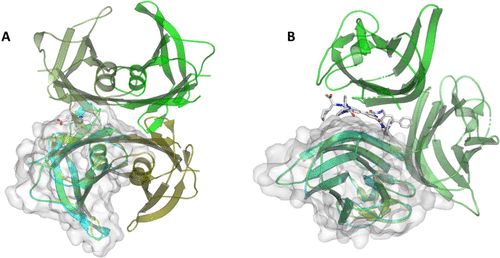
图 6. (A) Tafamadis (灰色棒) 与运甲状腺素蛋白 ( 5KCV )的四聚体结构形式结合不同深浅的绿色单体)。AF2 (青色卡通和表面) 确实准确地预测了结构的一个单体。(B) 靶向同源三聚体 CD40L 复合物 ( 3LKJ,不同深浅的绿色单体)。该化合物被锁定在由三个单体形成的裂缝中。根据单体AF2结构 (青色卡通和表面),无法预测此口袋。 CD40L是一种肿瘤坏死因子,主要在活化的T细胞表面表达。一些针对CD40L及其相互作用的抗体已经进入临床,用于治疗狼疮性肾炎、异体胰岛移植排斥反应和动脉硬化。文献中报道了一种抑制CD40L的小分子BIO8898的晶体结构,如图6B所示。引人注目的是,该分子没有结合到蛋白复合物的表面,而是深埋在亚单位之间,改变了蛋白与蛋白之间的相互作用及其对称性。目前仅能预测单个氨基酸链。进一步推进算法,以训练和预测一个结构是否以多聚体形式存在,是AF2发布以来已经做出的改进之一。然而,预测界面仍然具有挑战性,不能像预测单一蛋白质链那样准确和肯定地进行。
瞄准蛋白质-DNA和蛋白质-RNA界面是具有挑战性的,改变蛋白质-DNA结合特性的药物处于癌症治疗的第一线。 转录因子是引人注目的肿瘤学靶点,因为可以利用癌细胞的基因表达改变来进行治疗。转录可以通过小的DNA结合化合物,通过抑制蛋白质-蛋白质的相互作用或通过与转录因子的DNA结合域的结合而成为靶点。后者长期以来被认为是不可药用的。Huang等人报道了直接与STAT3的DNA结合域结合的分子,抑制了其转录活性,是该类药物的概念证明。 **AF2 在预测域间联系方面的能力不如域内联系好,这突出了未来版本改进的可能途径。**此外,AF2 可以在一定程度上了解蛋白质-蛋白质、蛋白质-DNA和蛋白质-RNA 结构域的情况,就像PDB中不同复杂类型的示例一样。样本数量相当有限,这使得ML算法对这些结构特征的预测变得复杂。学习准确可靠地预测这些更具挑战性的结构是未来的目标之一,直到我们可以声称蛋白质结构预测问题得到解决。
预测蛋白质配体复合物
AF2的预测是纯粹基于蛋白质的氨基酸序列的。获得蛋白质在其载脂状态 (apo state) 下的准确结构对每个药物设计项目来说都是一个巨大的突破,因为它可以在蛋白质结构上寻找可能是可药用的口袋。在确定了一个口袋后,计算模型可以帮助选择适合口袋形状的小分子。然而,这是对蛋白质配体结合过程的一种简化。其基本假设是,蛋白质配体结合是以锁 (蛋白质) 和钥匙 (配体) 的方式进行。事实证明,这种假设在大多数情况下是过度简化了。 如今,蛋白质配体结合可以通过两个互补的模型来理解。首先,一个蛋白质有多种构象,这些构象可能与晶体结构有很大的不同,化合物有选择地与这些构象中的一个结合。第二,诱导拟合,描述蛋白质口袋形状可能发生变化以适应配体进入其口袋的过程。 图7显示了谷氨酸电离受体AMPA型亚单位2 (GLUR2) 的一个例子。GLUR2是在人类AMPA受体中发现的一个亚单位,是一个已知的癫痫分子靶点。GLUR2的无配体结构 (PDB代码1FTO) 和配体结合结构 (PDB代码1FTM) 之间的差异相当大,例如,残基GLU193被翻转,相应的环被移动了5埃。AF2从这两个结构中学习了一些特征,因此,预测的结构是apo和holo蛋白的混合形式,这与在激酶中观察到的活性和非活性构象的行为相似 (上文MAPK1的例子,图3C)。如上所述,如果能有独立预测apo和holo状态的模型将是有益的。
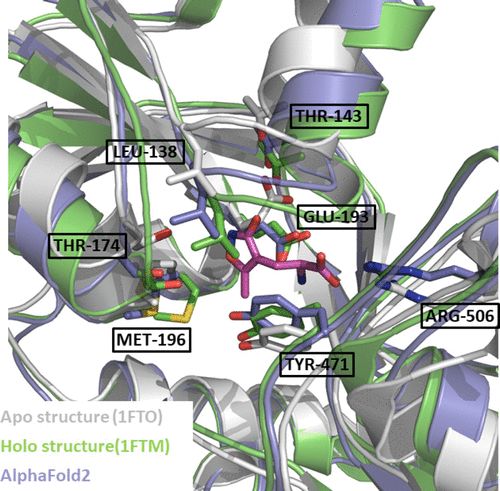
图7. 谷氨酸电离受体AMPA型亚单位2在其apo (灰色) 和holo (绿色) 形式下的蛋白质结构。来自AF2的预测结构以蓝色显示。AF2类似于实验中已知的apo和holo结构的混合物。 蛋白质的动态变化以及配体的动态变化 (在较小程度上) 使得蛋白质配体结合成为一个更难准确预测的问题。与独立的蛋白质结构的apo预测相比,这个问题增加了多层复杂性。与蛋白质由20个氨基酸组成相比,小分子可以由几乎无穷无尽的不同结构块生成。蛋白质和配体往往不是相互共价结合的。
预测小分子配体的结合位置是一项艰巨的任务,在药物发现中还没有得到解决 (对****接问题)。然而,将现有的小分子对接方法与精确的结构预测能力相结合,可以改善计算药物发现。
接下来,更具挑战性的是估计一个配体可能与某个口袋结合的强度 (评分问题)。这是药物发现的圣杯,已有多种方法来描述蛋白质配体的结合,其准确性各不相同。使用基于结构的对接的虚拟筛选试图用一个相对简单的能量函数来估计一个化合物的结合能。它通常不考虑蛋白质灵活性的能量学,如果它考虑的话,通常也非常有限。自由能计算通常是更准确的预测,因为它们考虑了蛋白质和配体的灵活性。然而,与虚拟筛选相比,要求进行广泛的计算,而且一次只能筛选少量的两位数的化合物。 当使用像虚拟筛选这样的方法时,侧链往往被固定在其初始位置。因此,**预测侧链在口袋中的方向变得比全局晶体结构预测更重要。**如第2节所述,AF2和RoseTTAFold的改进之一是端到端结构预测,这增加了侧链的准确性。根据蛋白质结构的不同,这种提高的准确性也是不同的。DeepMind在Nature杂志论文中所示的例子中,侧链的准确性令人印象深刻。然而,对于其他蛋白质来说,侧链的建模并不那么准确。Jumper等人已经强调,对于plDDT得分高的氨基酸,侧链的准确性更可能是正确的。与骨架结构预测相比,AF2对其猜测的准确度进行了预测,而对侧链的预测则没有这个功能。因此,不容易确定侧链是否以高置信度被预测。 不同的侧链灵活性是基于结构的药物设计项目的一个问题。**在当前版本的AF2中,侧链方向只是一个次要的方面,仅将其纳入辅助损失。**将侧链方向作为此类方法的主要目标,会对AF2结构如何用于药物设计产生很大影响。
进一步的复杂性是,蛋白质配体的过程是在水环境中进行的。水分子通过介导蛋白质和配体之间的相互作用,在蛋白质配体结合中起着关键作用。在对接过程和分子动力学模拟中,经常保留晶体中的水分子。**AF2被设计为仅预测蛋白质结构,因此目前没有预测任何水的位置。**然而,PDB中关于水分子的信息是多方面的。准确预测晶体结构中的水分子,甚至进一步预测哪些水参与配体的相互作用,是未来结构预测工具的另一个挑战。水的位置可以通过另一种深度学习方法从PDB中的结晶水中学习,或者通过应用基于物理学的方法,例如WaterMap, GIST, 3DRISM和SZMAP来预测蛋白质结构。
基于人工智能的蛋白质稳定性方法
单点突变在蛋白质中频繁发生,是导致大量遗传疾病的原因,也是导致多种癌症的原因。单个突变能够导致蛋白质失去其结构稳定性,并从其原始状态展开。 p53的多个突变能导致蛋白质结构的改变,这与它的功能丧失有关。p53的突变要么引起p53-DNA相互作用的构象变化,要么引起更全面的结构变化,降低p53的热力学稳定性。 最近报道了几种针对肿瘤抑制因子p53突变的候选药物。它们要么是恢复野生型的功能,要么是降解突变版的蛋白。多个候选药物,包括小分子药物,但也包括抗体,现在正在进行临床开发。**所有的ML方法都是在折叠结构上训练的,因此偏向于预测一个折叠的蛋白质结构。因此,导致蛋白质展开的突变不太可能被正确预测。**此外,导致蛋白质结构发生较小变化的突变,仍然会对蛋白质的功能产生严重影响,也很难像药物发现所需的那样准确预测。 几十种抗体被用于诊断和治疗疾病。今天,大多数临床使用的抗体都来自于自然界,而不是计算建模。计算方法大多集中在具有高二级结构含量和理想侧链相互作用的稳定蛋白质上。然而,蛋白质的分子功能往往需要长环和空腔,这是对整个蛋白质结构的不稳定。因此,预测具有这种长环的蛋白质的稳定性是一个挑战,正如在抗体的可变片段中看到的那样。 遗憾的是,正如Ivankov和他的同事所强调的,**对蛋白质稳定性的准确预测并没有在当前版本的结构预测中实现。**然而,存在几种预测蛋白质稳定性的计算方法。将新的结构预测工具的准确结构预测与预测蛋白质稳定性的能力配对,似乎是这些方法的一个有趣的机会,将有助于抗体的设计,同时也有助于揭示导致不稳定突变的疾病。 结语 AF2在最新的CASP挑战中表现出令人印象深刻的准确性,这在以前是从来没有过的。然而,我们相信这只是基于人工智能的方法促进解决结构预测问题的一个起点。由于前面提到的所有挑战,在我们宣布蛋白质结构预测挑战已经成功解决之前,还有很多额外的工作要做。 尽管如此,目前的进步还是令人印象深刻。**本文强调了现代结构预测工具的优势和劣势,以及它们如何已经用于药物设计。通过了解当前方法的缺点,可以更容易地将这些工具用于药物设计过程中。**到目前为止,结构预测工具只是用来增加从实验获得的结构知识。在没有实验证据的情况下,纯粹从模型结构开始药物设计工作,现在可能还很牵强。然而,如果这些模型使用得当,并能产生有用的预测结果,就可以建立对预测结果的信任。 此外,多个研究小组开始解决AF2的一些缺点,如多聚体预测、多构象等。RoseTTAFold和AF2现在都是开源的,这使得社区的其他成员能够在他们令人印象深刻的工作基础上进一步发展ML系统,以解决仍然摆在我们面前的额外挑战,并帮助药物设计加速。我们相信,基于人工智能的蛋白质预测和设计方法,随着它们的不断成熟,将在生物学和医学中发挥越来越重要的作用。 参考资料 Schauperl M, Denny RA. AI-Based Protein Structure Prediction in Drug Discovery: Impacts and Challenges. J Chem Inf Model. 2022 Jun 21. doi: 10.1021/acs.jcim.2c00026.
--------- End ---------