珍藏版:15000字深度长文,详解人类癌症与免疫治疗发展史

人与人之间的基因组相似度在99.99%。我们血缘关系最近的亲属——黑猩猩,在基因上与我们人类的相似度达到了96%。人类和猕猴共有93%的DNA。猫的基因与我们人类有着90%的相似性。
一、免疫治疗简史

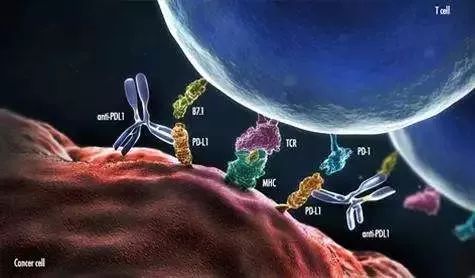
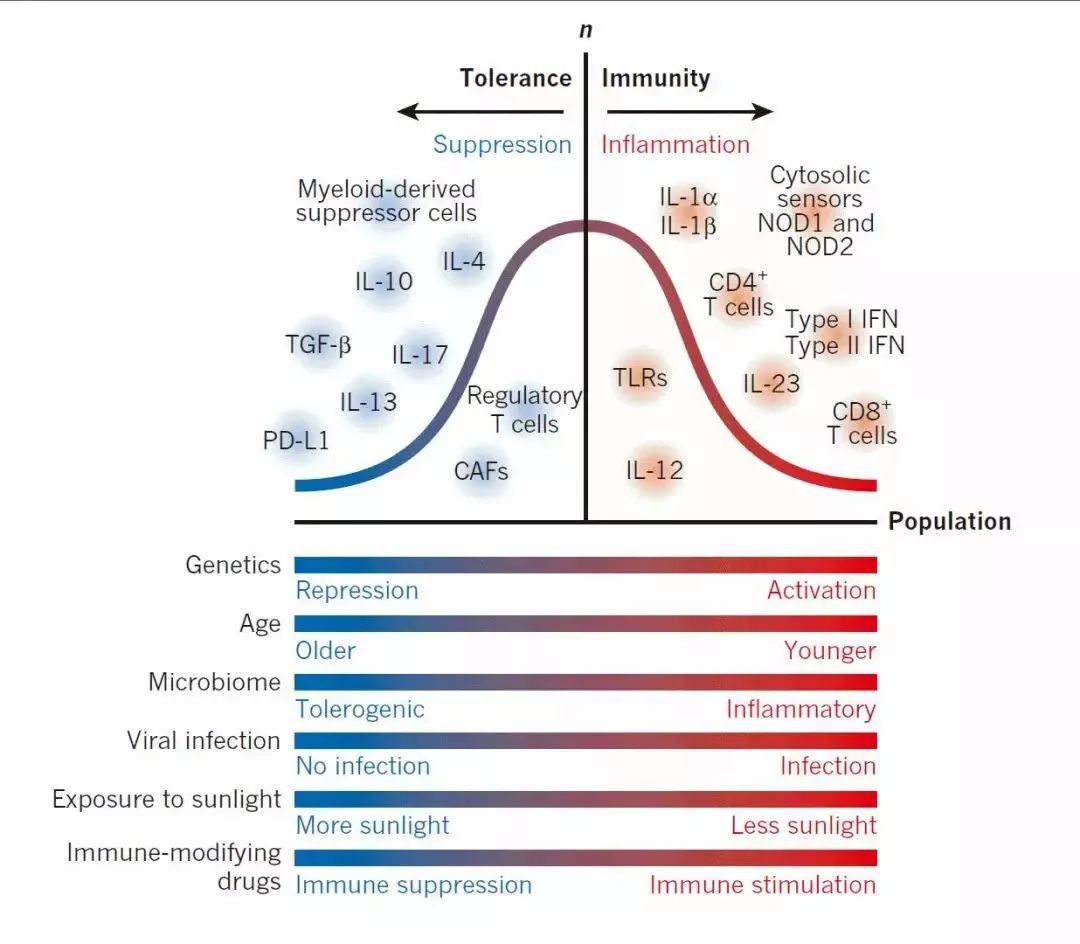
二、抗癌免疫学的基本原理

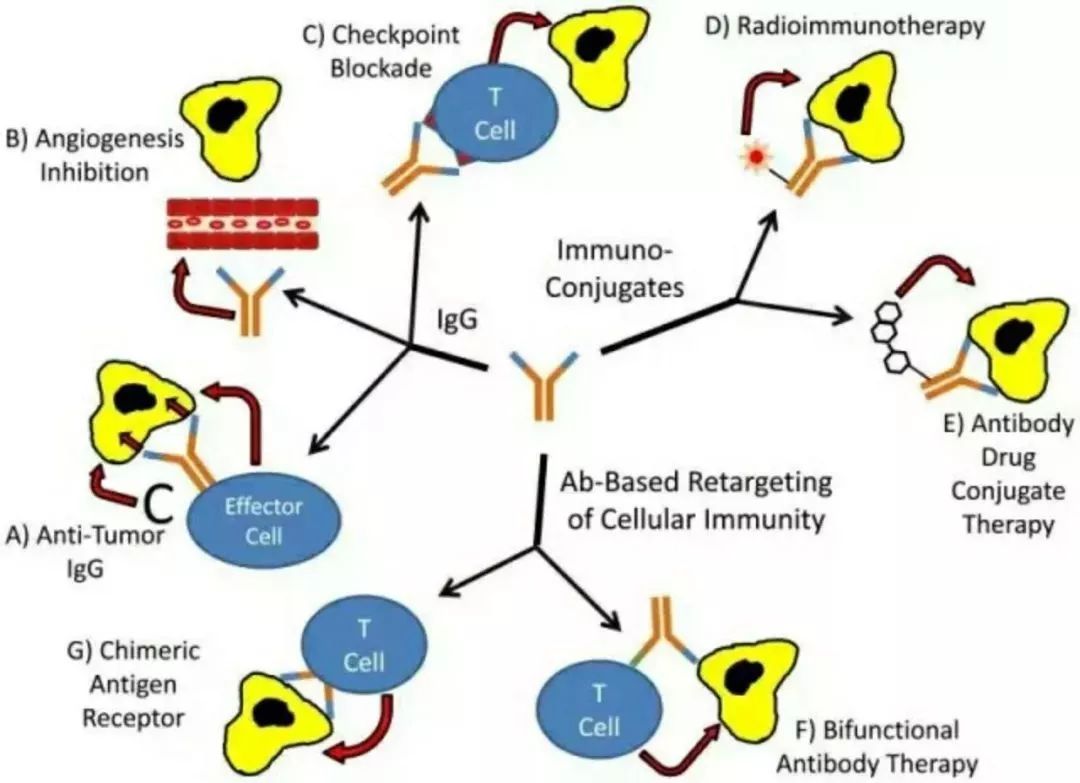
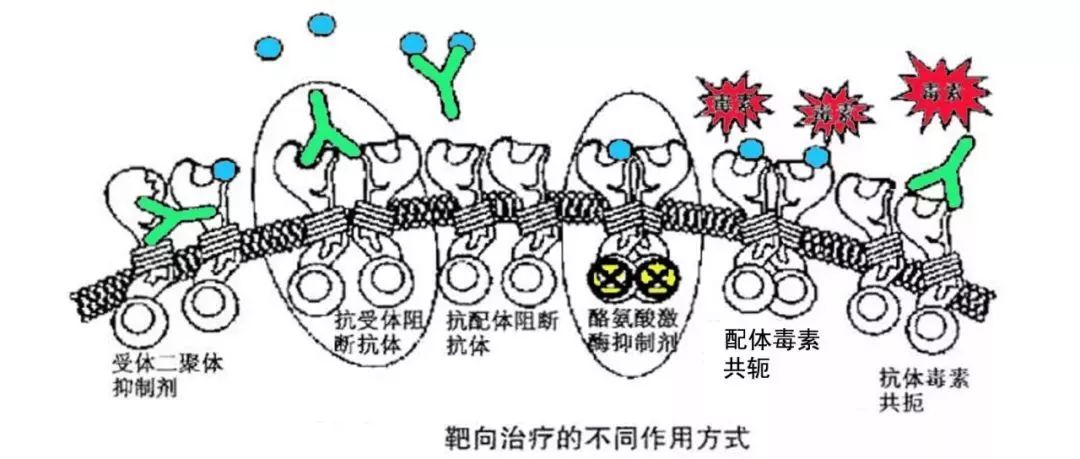
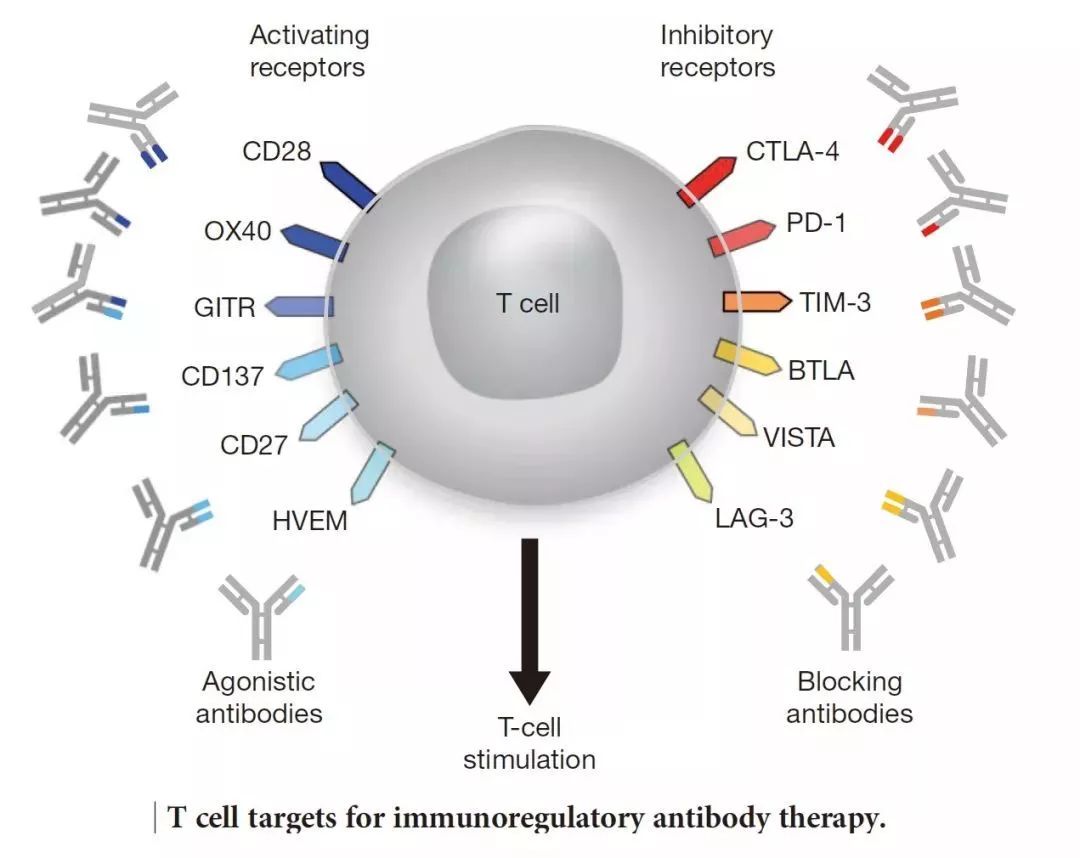
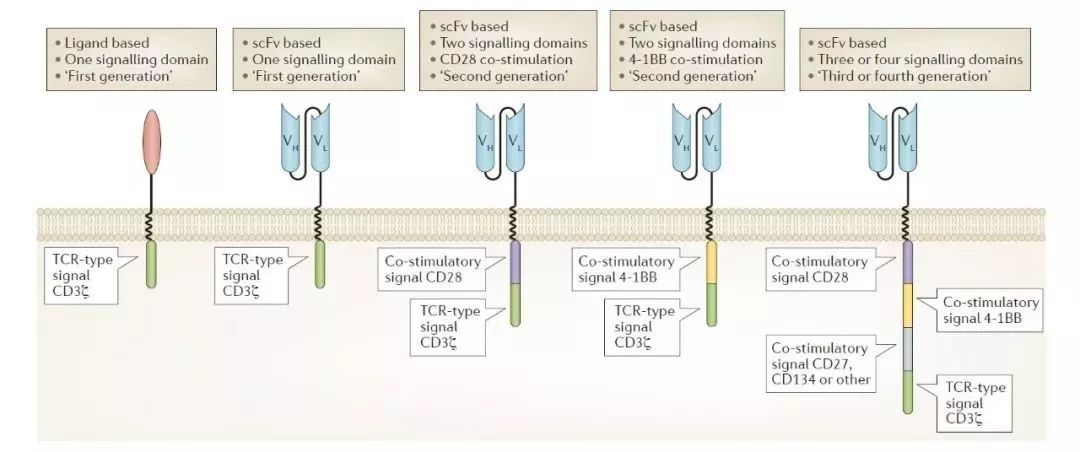
三、主要抗癌免疫疗法
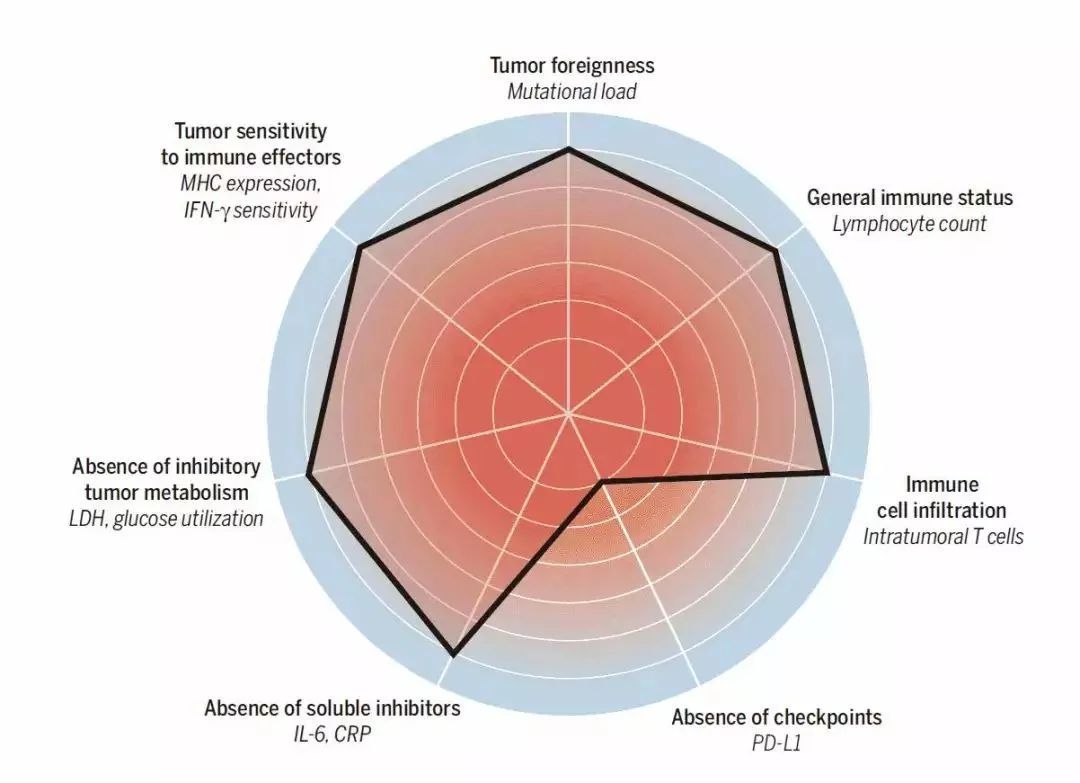
四、免疫评估
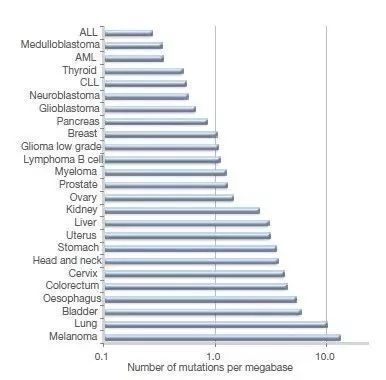
五、毒性管理与疗效评价

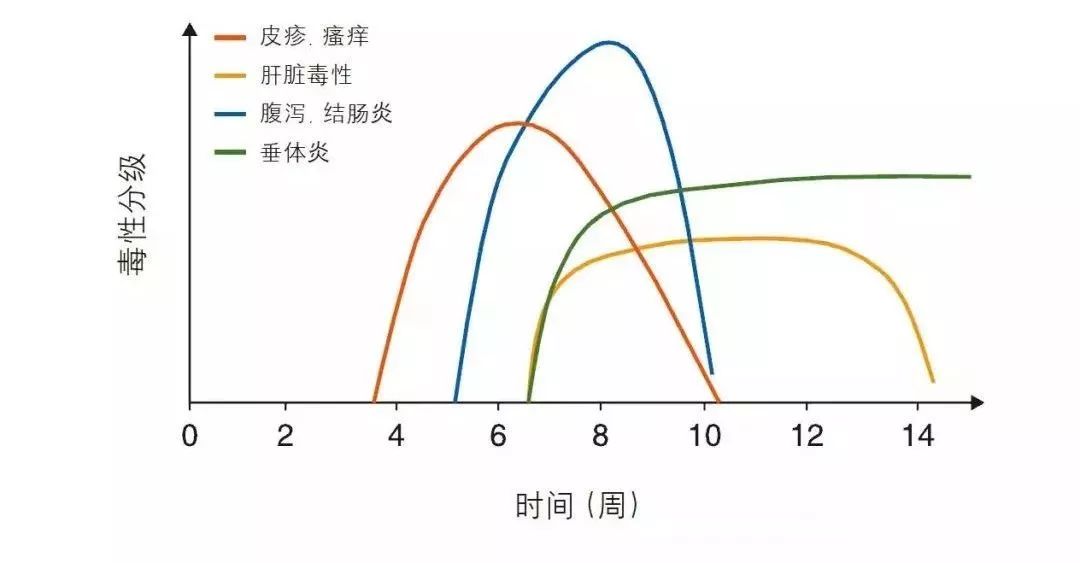
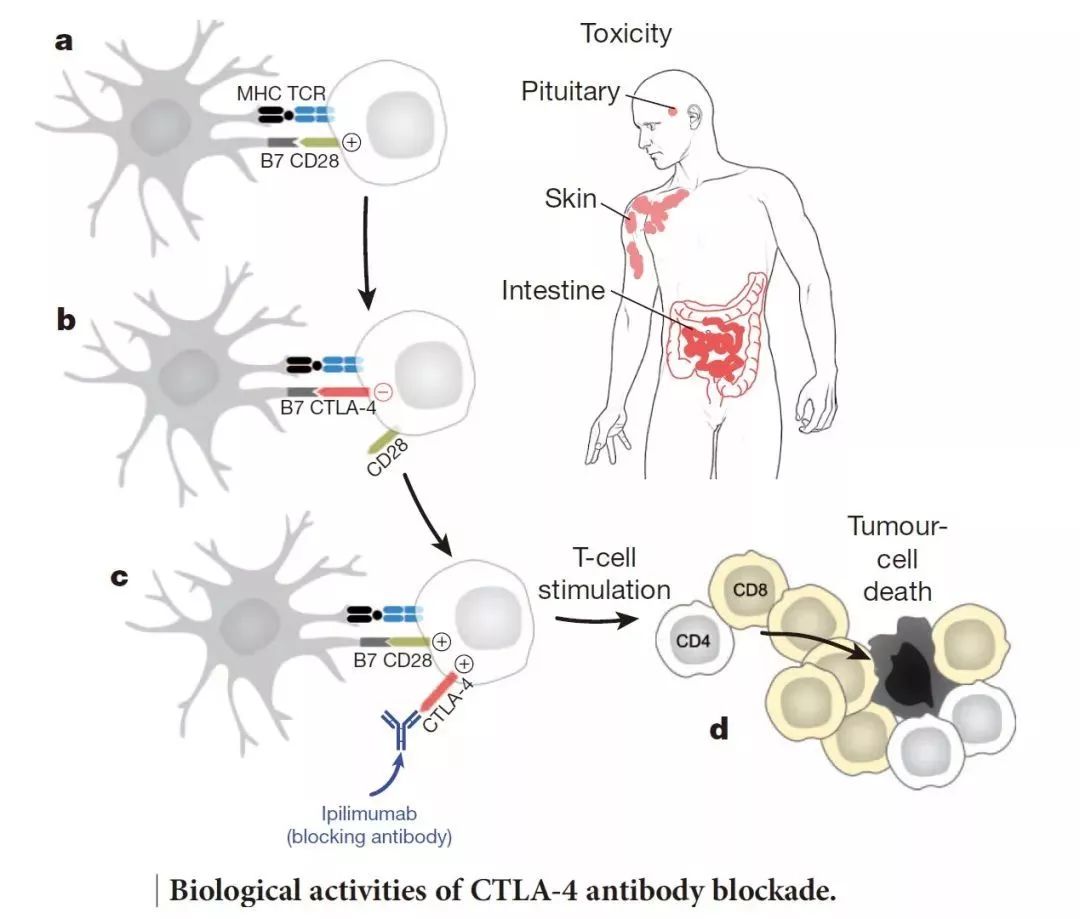
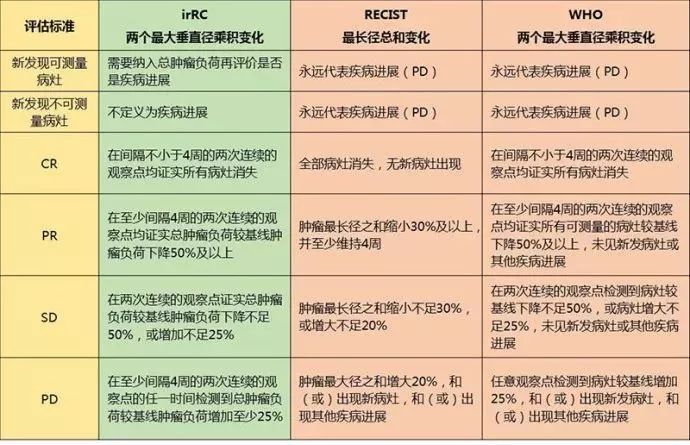
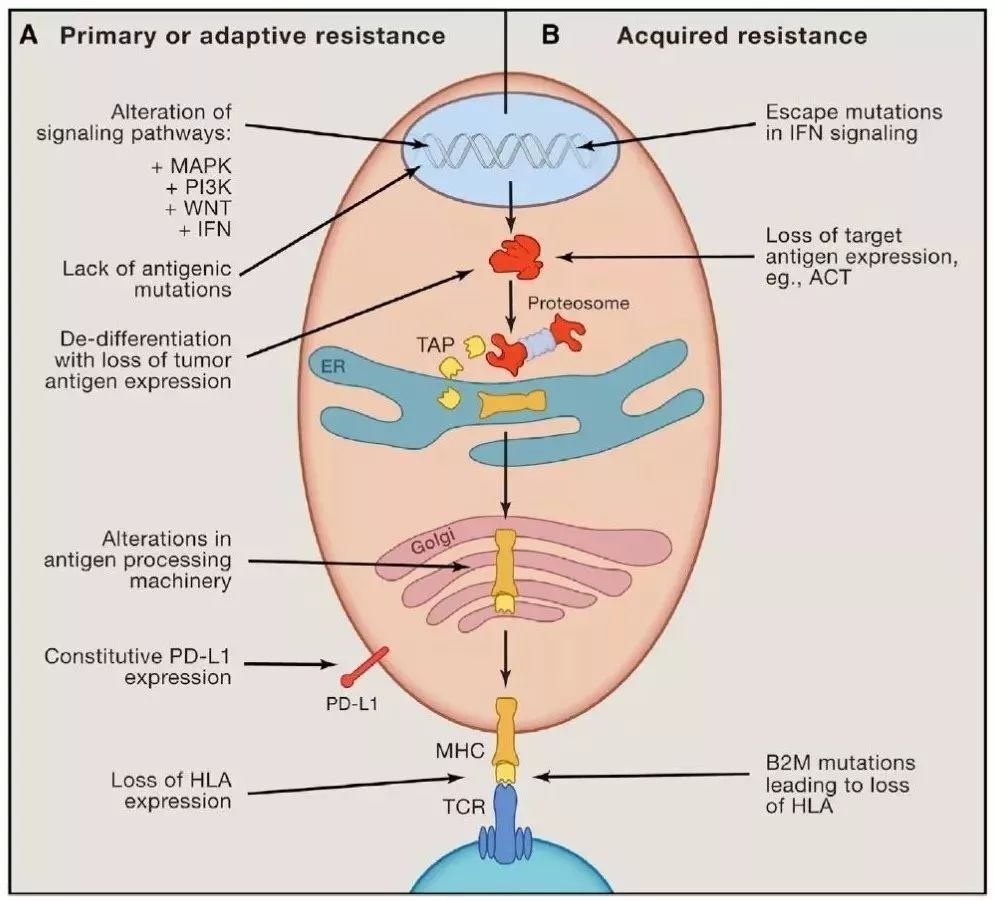
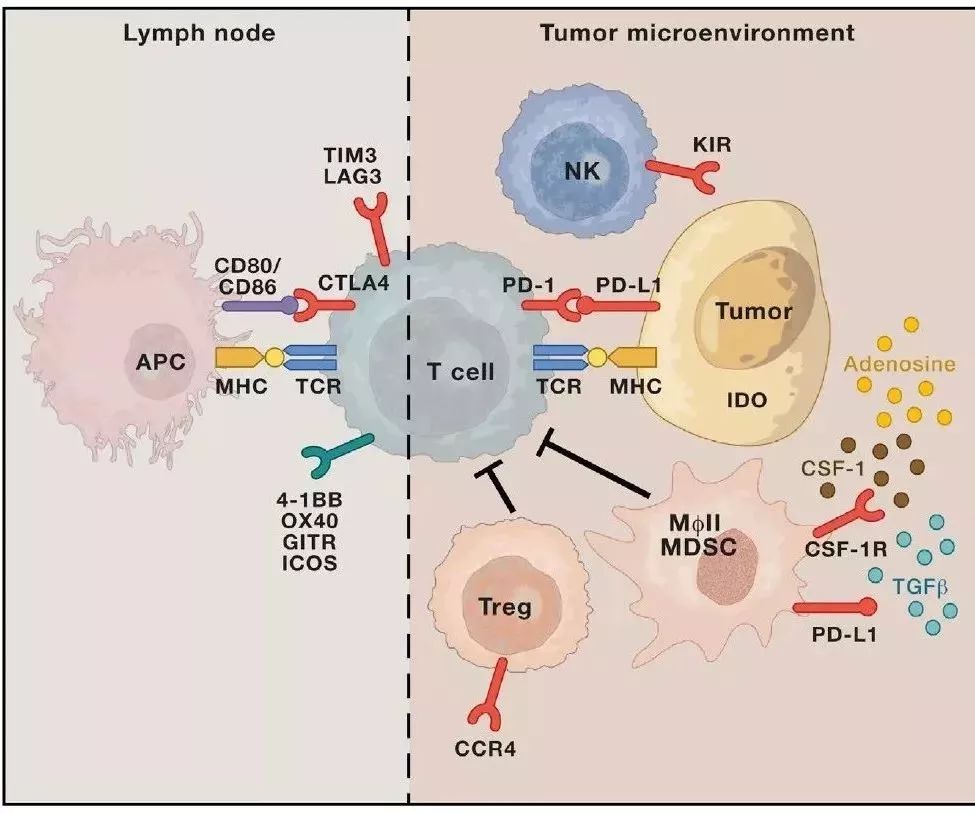
六、ICIs耐药现象的解释
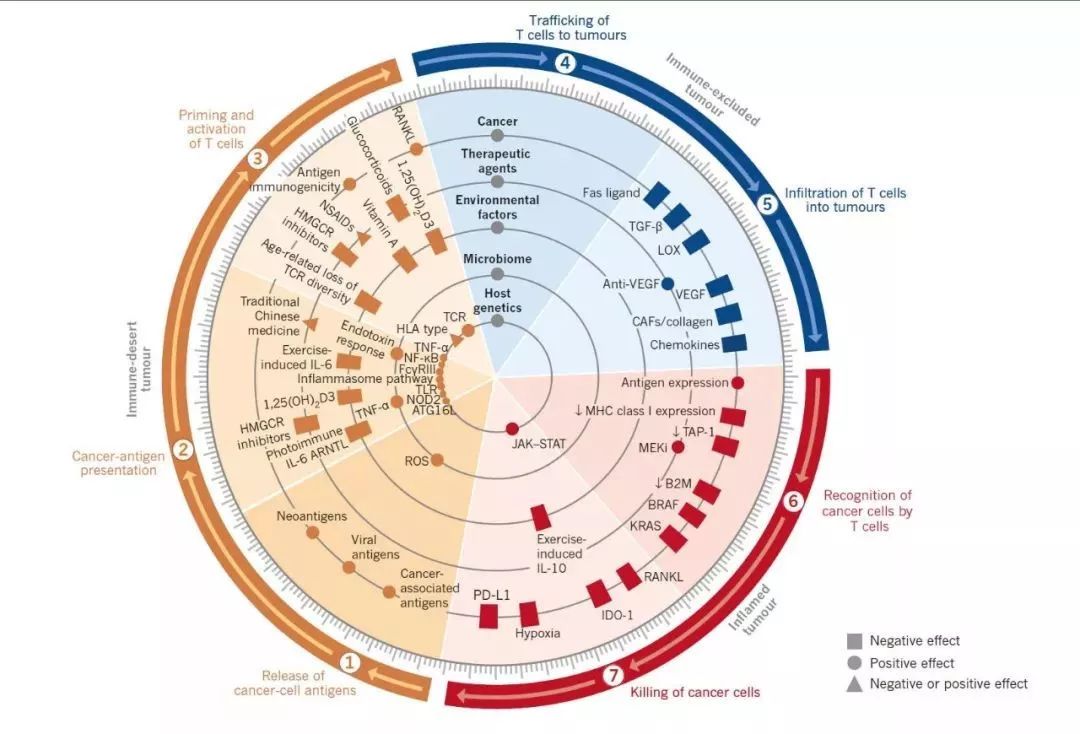
七、临床应用
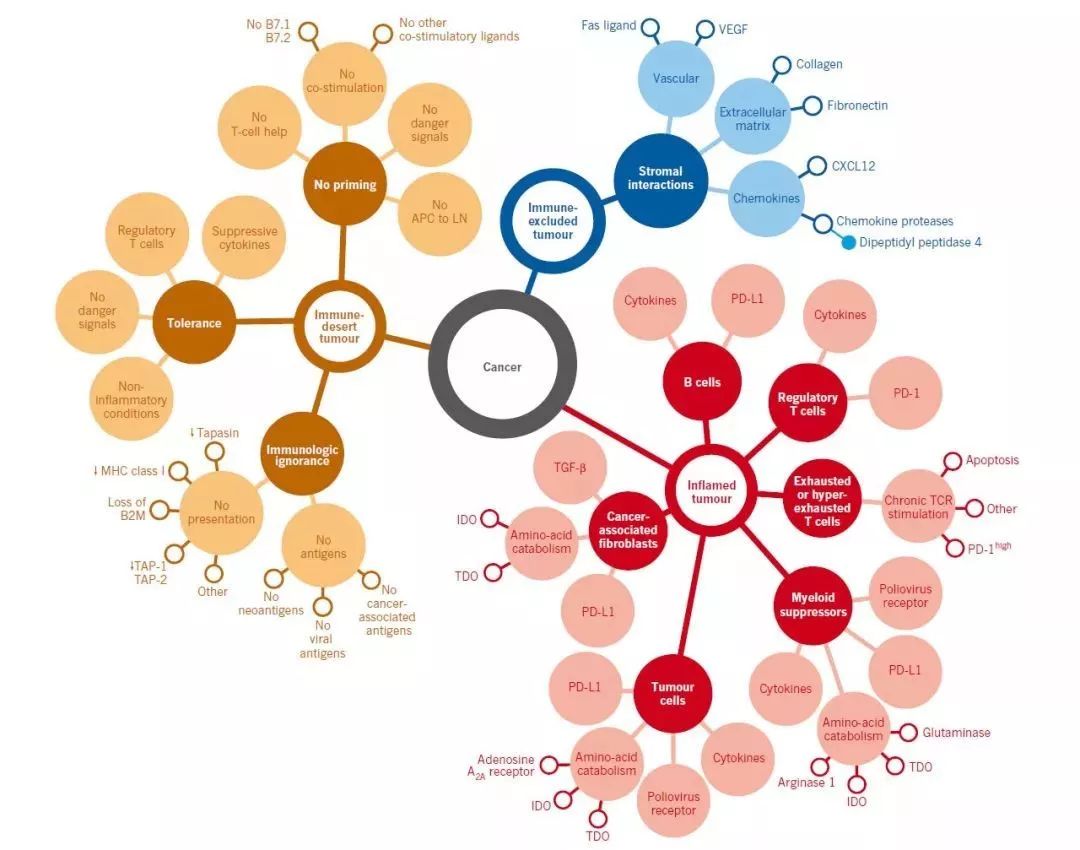








登录查看更多
相关内容
Arxiv
6+阅读 · 2018年7月19日



相关VIP内容
相关资讯
相关论文
Arxiv
6+阅读 · 2018年7月19日